胰岛β细胞去分化与2型糖尿病*
2024-04-07张茂森章卫平
张茂森, 张 海, 章卫平
(海军军医大学病理生理学教研室,上海 200433)
最新的流行病学数据显示,截止2018 年,我国糖尿病患病率已上升至12.4%,在非传染性疾病中仅次于心血管疾病和肿瘤,是严重威胁我国人民健康的常见病、多发病[1],其中绝大多数为2 型糖尿病(type 2 diabetes, T2D)。胰岛β 细胞数量减少与胰岛素分泌功能障碍是T2D 的病理生理学基础之一。以往认为,β 细胞减少与细胞凋亡相关,近十年的研究发现β 细胞去分化是T2D 进程中β 细胞衰竭的主要原因。本文就β 细胞去分化的特征、原因和机制等最新研究进展做一综述,为更深入地了解T2D 的发病机制和干预方法提供参考。
1 β细胞分化发育与去分化
1.1 β细胞分化发育的转录调控 胰岛存在多种内分泌细胞,包括α 细胞、β 细胞、δ 细胞和PP 细胞等,它们分别产生和分泌胰高血糖素、胰岛素、生长抑素和胰多肽等,共同调节机体的血糖稳态[2]。目前对β细胞分化发育的认识主要来自于啮齿类动物的实验研究。研究表明在一系列转录因子的调控下,内分泌祖细胞分化发育成为内分泌前体细胞,最终发育为成熟的β细胞[3]。
在内分泌祖细胞分化发育阶段,胰十二指肠同源盒基因(pancreatic and duodenal homeobox 1,Pdx1)、胰腺特异转录因子1a(pancreas-specific tran‐scription factor 1a, Ptf1a)和性别决定区Y 框蛋白9(sex determining region Y-box 9, Sox9)等转录因子开始表达并发挥了重要的调控作用[3]。其中Pdx1 可促进胰腺的早期发育,且可促进祖细胞向内分泌前体细胞分化,β 细胞敲除Pdx1可造成小鼠胚胎腺泡和β 细胞的分化障碍,导致胰腺发育不全[4]。同样的,Sox9 也参与胰腺早期内分泌祖细胞的增殖与分化,是胰腺内分泌发育过程调控网络中的多潜能因子。敲除小鼠β 细胞Sox9基因发现胰腺早期发育障碍[5]。Ptf1a的表达在Pdx1和Sox9之后,且Ptf1a的正常表达受Pdx1 和Sox9 的协同调控。Ptf1a基因敲除后可导致小鼠胰腺发育不全,且分化方向由胰腺祖细胞向十二指肠谱系发育[6]。
在内分泌前体细胞分化阶段,主要以Pdx1、神经元 素3(neurogenin 3, Ngn3)、配对盒转录因子4(paired box 4, Pax4)和叉头盒蛋白O1(forkhead box O1, FoxO1)等转录因子发挥了重要的作用,此阶段内分泌前体细胞具有分化为各类内分泌细胞的潜能[3]。其中Ngn3是胰腺祖细胞向内分泌前体细胞转化所必需的激活因子,胰腺内分泌细胞的发育均需要Ngn3 的调控[7]。而Pax4 是β 细胞和δ 细胞的发育及胰岛素产生所必需的转录因子。缺乏Pax4的小鼠胚胎胰腺不能分化出β 细胞[8]。FoxO1 可以与Notch 信号相互作用调控分化关键转录因子Ngn3 的表达,从而调节胰腺内分泌细胞的分化[9]。
在最后发育成熟阶段,β 细胞主要以Pdx1、MafA(musculoaponeurotic fibrosarcoma oncogene homolog A)、Nkx6.1(NK6 homeobox 1)、NeuroD(neurogenic dif‐ferentiation)和Ins等基因的表达为特征[3]。研究表明,MafA 不仅是胰岛素生物合成和葡萄糖刺激的胰岛素分泌(glucose-stimulated insulin secretion, GSIS)的关键调节因子,而且对于维持β 细胞的成熟表型也是必不可少,MafA缺失的小鼠胰腺中β 细胞关键的特征性基因(如Ins1、Pdx1和NeuroD等)表达下调,小鼠胰岛素合成障碍、糖耐量受损且胰岛形态异常[10]。在Nkx6.1 的调控下Pdx1 与Ngn3 双阳性的前体细胞可转化为胰岛β 细胞。Nkx6.1基因敲除小鼠胰腺中未发现成熟的β 细胞[11]。NeuroD 是参与内分泌细胞谱系发育的碱性环-螺旋(basic loop-helix)转录因子,并可以启动Ins基因的表达,在小鼠β 细胞敲除NeuroD后,小鼠出现胰岛素合成障碍和糖耐量受损[12]。
综上,Pdx1、Sox9、Ptf1a、Ngn3、Nkx6.1、MafA、NeuroD 等转录因子不仅在β 细胞的发育、分化成熟方面发挥作用,而且在维持β 细胞的特征和功能上也发挥重要作用(图1)。有研究表明,啮齿类动物和人类的内分泌细胞发育过程大体一致,个别发育阶段转录因子的表达仅有小部分不同。如在啮齿动物中,β 细胞分化成熟过程中,有MafB 到MafA 表达的变化,而在人类成熟的β细胞中保持MafB表达[13]。
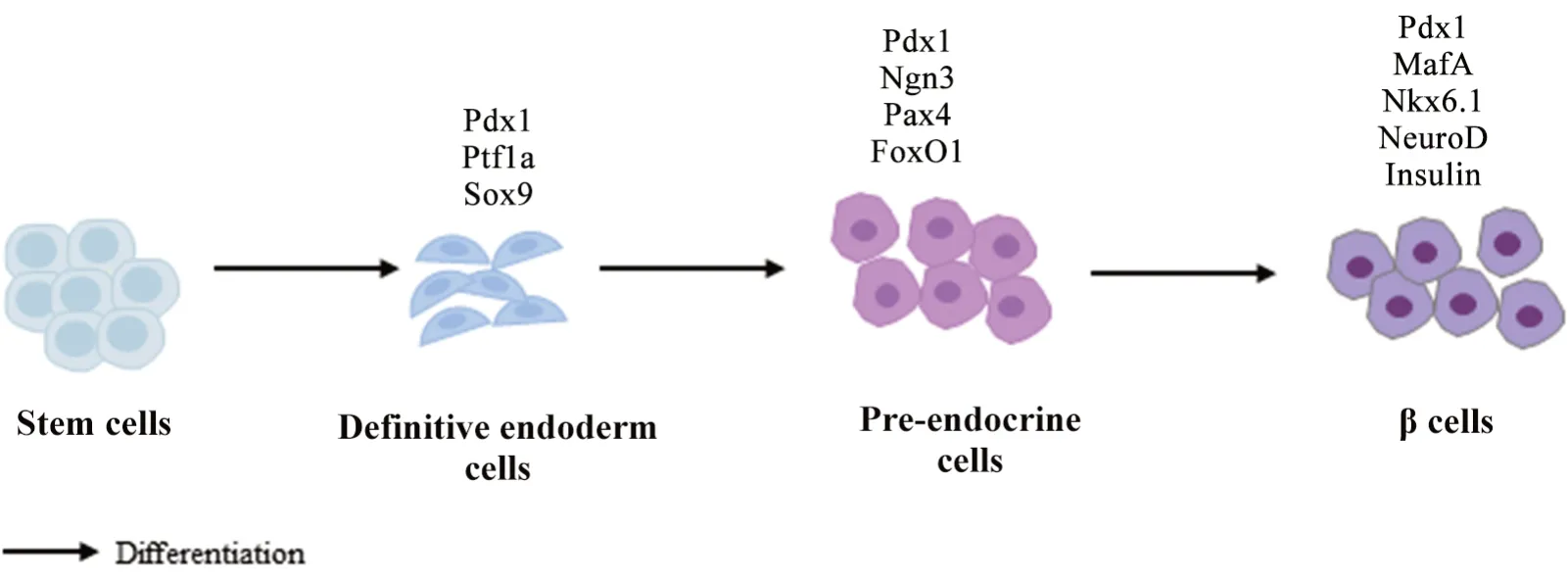
Figure 1. Schematic diagram of islet β cell diffetentiation and development. Pdx1: pancreatic and duodenal homeobox 1; Ptf1a:pancreas-specific transcription factor 1a; Sox9: sex determining region Y-box 9; Ngn3: neurogenin 3; Pax4: paired box 4; FoxO1: forkhead box O1; MafA: musculoaponeurotic fibrosarcoma oncogene homolog A; Nkx6.1: NK6 homeobox 1;NeuroD: neurogenic differentiation.图1 胰岛β细胞分化发育示意图
1.2 β 细胞去分化的特征性变化 2012 年,哥伦比亚大学Accili 实验室利用细胞谱系示踪技术,在β 细胞特异性敲除FoxO1的糖尿病小鼠中发现β 细胞去分化的现象,首次提出并证实该模型小鼠中成熟β细胞数目减少的主要原因是β 细胞去分化而非细胞凋亡[14],随后在T2D 病人的胰岛中检测到β 细胞去分化[15]。Amo-Shiinoki 等[16]在T2D 患者中发现β 细胞数量减少,但β 细胞的凋亡率相对较低,同时观察到β 细胞分化表型发生了变化,主要为内分泌祖细胞标志物表达上调,提示β 细胞去分化为无胰岛素分泌功能的内分泌祖细胞。Dai 等[17]也发现慢性高血糖、高血脂和外周胰岛素抵抗并未导致移植后的β细胞发生凋亡,而是主要通过下调β 细胞的特征性转录因子、葡萄糖代谢基因和蛋白质分泌相关基因的表达,使其发生去分化。同时,内分泌祖细胞标志物及β细胞中禁止表达的基因(forbidden genes)表达上调,从而损害β细胞分泌胰岛素的功能和减少β细胞数量。
近年来的研究表明,成熟的β 细胞特性不稳定,在一定的病理生理条件下(如高血糖、高血脂)β细胞会不同程度地失去其分化的表型和细胞特性发生去分化[14,17]。β细胞特性的维持受许多因素的调节,包括转录因子和染色质修饰[18]。β 细胞去分化是指分化成熟的β 细胞转化为祖细胞样细胞或转分化为非β 细胞,如α 细胞、δ 细胞和PP 细胞。β 细胞去分化的主要特征包括:(1)β 细胞富集基因(β cell-en‐riched genes)表达下调,包括编码β 细胞特征和功能的转录因子、葡萄糖代谢相关、胰岛素合成、加工和分泌相关蛋白的基因;(2)内分泌祖细胞特征性相关基因表达上调;(3)β 细胞禁止基因(β cell-forbidden genes)表达上调。
目前的研究认为,β细胞去分化为无胰岛素分泌功能的内分泌祖细胞样细胞是导致T2D 患者β 细胞功能障碍的主要病理因素,是影响T2D 发生发展的关键环节[14]。因此,了解β 细胞去分化的原因和机制,可望为T2D 的预防和治疗提供新的策略和干预靶点。
2 β细胞去分化的原因
T2D 主要受遗传(如基因突变)和环境(如饮食)等多因素的影响。在对T2D 患者进行全基因组关联研究中分析发现,在东亚和美洲原住民人群中涉及溶质载体家族16 成员11(solute carrier family 16 member 11,SLC16A11)基因的突变以及在格陵兰岛因纽特人群中涉及TBC1 域家族成员446(Tre-2/BUB2/cdc1 domain family 446,TBC1D446)基因的突变可增加患T2D 的风险[19]。其中SLC16A11基因突变导致肝脏中SLC16A11的mRNA 表达水平降低,使肝细胞表面转运蛋白水平降低,导致脂肪酸堆积和脂质代谢紊乱进而引发T2D[20]。TBC1D446 参与肌肉骨骼肌细胞葡萄糖转运蛋白4(glucose transporter 4, Glut4)在细胞内的转位过程,可调节胰岛素刺激的骨骼肌细胞葡萄糖转运,其突变可引起Glut4 转位异常导致骨骼肌细胞葡萄糖转运能力下降,葡萄糖的摄取减少使葡萄糖的代谢障碍导致血糖升高从而引发T2D,这些关键基因位点发生突变会使会使体内糖脂代谢发生障碍进而导致T2D[21]。对来自15名糖尿病和15 名非糖尿病器官捐赠者的胰岛分析发现,糖尿病患者β 细胞特征性转录因子(如NKX6.1)表达下调,而内分泌谱系标志物(如FoxO1)和内分泌祖细胞标志物[如乙醛脱氢酶1 家族成员A3(alde‐hyde dehydrogenase 1 family member A3, Aldh1a3)]表达上调,提示在T2D 患者中β 细胞发生了去分化[15]。
忽然,四周的笑声大了起来。我连忙朝后一看,只见巴克夏两手平端脸盆,站在“等腰三角形”的顶角上,正怪模怪样地走着,而我和她的手不到二十厘米的距离。稍远一些是一帮孩子跟在后面看新鲜。这种队形活像外国某电视剧中王子与公主去教堂举行婚礼。我顿时汗流浃背,不由加快了步子,把“等腰三角形”拉成了“不等边三角形”。
动物模型研究发现,在小鼠β 细胞中特异性敲除FoxO1,并不会导致β 细胞死亡,但β 细胞关键的特征性基因(如Pdx1、MafA和Nkx6.1等)表达下调,内分泌前体细胞标志物[如Ngn3、NeuroD1 和八聚体结合转录因子4(octamer-binding transcription factor 4, Oct4)等]表达增加,提示β 细胞发生去分化[14]。Dobosz 等[22]发现,小鼠β 细胞特异性敲除硬脂酰辅酶A 脱饱和酶1(stearoyl-coenzyme A desaturase 1,Scd1)基因后,胰岛的形态结构被破坏且β 细胞特征性基因Pdx1、Nkx6.1和MafA下调,β 细胞内分泌祖细胞标志物如Sox9 表达上调,提示β 细胞发生去分化。Sakano 等[23]在小鼠β 细胞中特异性敲除囊泡单胺转运蛋白2(vesicular monoamine transporter 2,Vmat2),发现β 细胞中的特征性基因(包括Ins1、Ins2、Glut2、Pdx1、Nkx6.1和MafA)表达下调,β 细胞内分泌祖细胞标志物(如Aldh1a3)的表达上调,表明β细胞发生去分化。综上,人群和模式动物中的研究表明,β细胞基因功能障碍可能引起去分化。
现代社会饮食中糖和脂肪的含量激增,也是T2D 发生发展的重要病因。短期的高血糖和高血脂会刺激胰岛素的合成和释放,而机体长期暴露于高血糖和高血脂条件下会产生糖毒性和脂毒性,糖脂毒性可以导致氧化应激、内质网应激和炎性细胞因子释放等来触发β细胞去分化,β细胞特征性转录因子包括MafA和Pdx1表达下调,导致胰岛素合成和分泌障碍,从而损害胰岛β 细胞功能,加快T2D 的发生发展[24]。在糖尿病患者胰岛中发现,在持续的高浓度葡萄糖刺激下β 细胞特征性转录因子(如Pdx1 和Nkx6.1等)表达下调,而内分泌祖细胞标志物(如Al‐dh1a3)表达上调提示β 细胞发生去分化[15]。有研究表明,对db/db糖尿病模型小鼠进行长期热量限制的干预使其保持正常血糖水平,可使其β 细胞特征性的转录因子(如Pdx1、Glut2、MafA 和Nkx6.1)的表达上调,提示限制热量摄入可以阻止β细胞去分化[25]。
在体外实验中,Neelankal 等[26]利用高糖培养基(22.5 mmol/L 葡萄糖)培养小鼠胰岛β 细胞株Min6,培养4 d 后分析发现β 细胞特征性的转录因子(如Pdx1 和MafA)表达下调,而内分泌祖细胞标志物(如Ngn3)表达上调,提示β 细胞发生去分化,但没有观察到大量β 细胞死亡。Kjørholt 等[27]利用25 mmol/L葡萄糖刺激分离的db/db小鼠胰岛,β 细胞成熟和功能相关基因(如Nkx6.1、NeuroD、Pdx1和Pax6)的表达下调,提示β 细胞发生去分化。以上均表明高糖是胰岛β细胞去分化的重要原因,这也是糖毒性对β细胞的病理作用。
脂毒性也可以引起β 细胞去分化和功能障碍。小鼠喂养高脂饮食13周后胰岛β细胞的胰岛素分泌功能障碍,内分泌祖细胞标志物Aldh1a3 的表达上调,提示β 细胞发生去分化[28]。Hagman 等[29]将大鼠的胰岛细胞分离培养并给予24.48 mmol/L 棕榈酸刺激5 h 发现,在高脂刺激下β 细胞特征性转录因子(如Pdx1、MafA 等)表达下调,提示β 细胞发生去分化。Staaf 等[30]分离人胰岛进行体外培养并给予3.0 mmol/L 棕榈酸刺激(产生脂毒性),分析发现棕榈酸刺激后胰岛β 细胞分泌胰岛素功能缺失;同时对肥胖人群和瘦弱人群进行空腹棕榈酸水平检测,发现肥胖受试者的棕榈酸平均水平更高。T2D 患者在减肥手术后的短期和中期可显著改善体重、血糖控制、血脂和β 细胞功能,这表明在消除高脂血症后,β 细胞功能恢复[31]。以上均表明高脂是胰岛β 细胞去分化的重要原因,这也是脂毒性对β细胞的病理作用。
综上所述,遗传因素(如基因突变)和环境(如饮食)因素都可使β 细胞发生去分化,促进T2D 的疾病进展。因此,进一步了解β 细胞去分化的机制,可以为初期干预β 细胞去分化或者诱导去分化细胞再分化以恢复β 细胞的功能提供方向,为临床治疗糖尿病提供新的思路。
3 β细胞去分化的机制
3.1 氧化应激 β 细胞分泌胰岛素需要ATP 的参与,ATP 主要来自葡萄糖代谢,其代谢过程容易产生大量的活性氧(reactive oxygen species, ROS)。细胞代谢产生ROS [如NADPH 氧化酶(NADPH oxidase,NOX)、过氧亚硝基阴离子(peroxynitrite, ONOO−)等],可在不同的亚细胞位置(如线粒体、内质网等)堆积[32]。对T2D 患者和糖尿病小鼠模型的胰岛研究发现,β细胞中高水平的葡萄糖代谢在线粒体中产生大量的ROS[33]。由于β 细胞中抗氧化酶水平较低且对ROS 特别敏感,使其容易因ROS 堆积导致氧化应激,从而损伤β 细胞功能[32]。长链游离脂肪酸(主要是棕榈酸)可在β 细胞氧化过程中产生过氧化氢[34]。由于β 细胞缺乏过氧化氢酶,过氧化氢的堆积可导致氧化应激。Leenders 等[32]在体外实验中用过氧化氢处理来自非糖尿病患者的原代胰岛细胞,导致活性氧的积累以诱发氧化应激,导致β 细胞特征性的转录因子包括MafA和Pdx1表达下调,而内分泌祖细胞标志物Sox9 的表达上调,提示在氧化应激条件下β 细胞发生去分化,β 细胞葡萄糖刺激的胰岛素分泌能力受损。将Ins1 细胞分别用高糖和含有棕榈酸的培养基培养以模拟糖毒性和脂毒性的刺激作用,ROS 的产生显著增加,并通过下调MafA 的表达影响胰岛素的合成从而减少胰岛素的分泌[35]。
此外,β 细胞的氧化应激还可以激活c-Jun 氨基末端激酶(c-Jun N-terminal kinase, JNK)通路,从而降低FoxO1 的磷酸化水平,进而抑制转录因子Pdx1的表达,使β细胞发生去分化导致功能障碍[36]。Latif等[37]在T2D 大鼠模型中发现,通过饮食或饮水补充多酚类的抗氧化剂可以增强β 细胞的氧自由基清除能力减少氧化应激,与患病组的β 细胞相比,补充抗氧化剂组的β 细胞特征性基因(如Pdx1、Ins1、Ngn3、Glut等)表达上调,从而促进β 细胞合成和分泌胰岛素。综上,氧化应激是β 细胞发生去分化的重要机制,安全有效的抗氧化剂可以为治疗T2D 提供新的选择。
3.2 炎症 研究发现T2D 患者胰岛的病理特征是免疫细胞、促炎细胞因子、趋化因子、细胞凋亡和淀粉样蛋白等大量沉积导致纤维化,这是胰腺β 细胞功能障碍重要原因之一[38-39]。其中促炎症细胞因子[如白细胞介素1β(interleukin-1β, IL-1β)、IL-6 和肿瘤坏死因子α(tumor necrosis factor-α, TNF-α)]已被证明可促进人和小鼠胰岛β 细胞的去分化。Wang等[40]利用非糖尿病胰腺导管腺癌患者的胰岛证明,在肿瘤微环境中无论是局部原位炎症还是肿瘤释放的体液细胞因子,均会导致β 细胞胰岛素分泌功能障碍,且内分泌祖细胞标志物Aldh1a3 的表达上调,表明仅炎症这一因素就可直接诱导β 细胞去分化。将小鼠胰岛暴露于非细胞毒性浓度的IL-1β 中,β 细胞特征基因(如MafA)的表达降低。去除IL-1β 后,β细胞特征基因恢复到刺激前的水平,β细胞功能恢复正常,表明炎性细胞因子刺激可导致β 细胞发生去分化[41]。Oshima 等[42]利用人β 细胞系EndoC-β H1模拟肠道感染中炎症因子对β 细胞的作用,发现炎症因子可使β 细胞特征性转录因子(如Pdx1 和Nkx6.1 等)表达下调,而内分泌祖细胞标志物(如Sox9)表达上调,从而提示β 细胞发生去分化。在此过程中NF-κB 通路的激活可能是β 细胞去分化的重要驱动力。Demine等[43]将诱导性多能干细胞诱导的β 细胞使用1 000 U/mL 的干扰素γ 和50 U/mL 的IL-1β 处理24 或48 h 后分析,发现β 细胞特征性转录因子(如Nkx2.2)表达下调,提示在炎症因子可使体外诱导分化的β 细胞发生去分化。目前对于炎症诱导β细胞去分化的机制尚不清楚,需要进一步的研究明确其机制,为治疗T2D提供新的干预方向。
在T2D 发生时,大量的内质网应激原持续刺激,如高血糖、高脂血症、低氧和促炎细胞因子(如TNFα、IL-1 等)会导致β 细胞分泌胰岛素功能障碍,同时发现β 细胞特征性转录因子(如Pdx1、MafA 和FoxO1)表达下调,而内分泌祖细胞标志物(如Ngn3和Oct4)表达上调,提示β 细胞发生去分化[45]。在内质网稳态即将失衡时,细胞会启动未折叠蛋白反应(unfolded protein response, UPR)作为细胞的一种保护性机制,UPR 可通过减少错误蛋白质的折叠来恢复内质网动态平衡[46]。内质网应激时UPR对于维持β 细胞的分化表型非常重要,UPR 的破坏可以导致β细胞特征性转录因子MafA 表达下调,使β 细胞发生去分化促进T2D 的进展[47]。许多因素如基因突变、细胞因子、感染、过量营养、胰岛淀粉样多肽和胰岛素抵抗等可以破坏胰腺β 细胞中UPR 平衡,从而触发内质网应激,最终导致β 细胞功能障碍[46]。总之,内质网应激可能是β细胞去分化的机制之一。
3.4 微小RNA(microRNA, miRNA, miR)和长链非编码RNA(long noncoding RNA, lncRNA) miRNA是一种内源性非编码小RNA,长19 到24 个核苷酸,通过与mRNA 的3´端非翻译区(untranslated region,UTR)特异性结合来介导RNA 沉默。目前报道多个miRNA 参与β 细胞的发育和功能成熟,如miR-204、miR-375、miR-7、miR-483 和miR-184、miR-200s 和miR-30s等。研究发现miR-204在人胰岛β细胞中可以直接下调胰腺β 细胞特征基因(如MafB)的表达,导致β 细胞发生去分化,胰岛素合成和分泌功能障碍[48]。在Min6细胞和原代胰岛中过表达miR-204可导致MafA、Pdx1 和NeuroD1 等β 细胞特征性转录因子表达下调,β细胞发生去分化[49]。Yongblah等[50]发现在小鼠胰岛中过表达miR-7 可以下调β 细胞特征性基因(如Pdx1、Pax6、Nkx6.1、NeuroD1和MafA)的表达,导致β 细胞发生去分化和胰岛素分泌功能受损。
目前发现胰腺β 细胞表达1 000 多个lncRNA。lncRNA 可以通过调节转录、控制mRNA 的降解来影响β 细胞胰岛素的产生和分泌、葡萄糖敏感性[51]。研究发现,β 细胞特异的lncRNA[如Pdx1 基因座上游转录物(Pdx1 locus upstream transcript, Pluto)、牛磺酸上调基因1(taurine up-regulated gene 1, Tug1)和母系表达基因3(maternally expressed gene 3, Meg3)等]缺陷会导致类糖尿病[52]。其中Akerman 等[53]发现Pluto 通过增强Pdx1启动子与其增强子簇之间的结合来促进Pdx1 表达,提示Pluto 在预防β 细胞去分化中发挥积极的作用。在体外Min6 细胞干扰Tug1和小鼠β 细胞Tug1敲除模型中发现β 细胞特征性基因(如Pdx1、NeuroD1和MafA)表达下调,提示Tug1与β 细胞去分化紧 密 相关[54]。在Min6 细胞干扰Meg3和小鼠β 细胞Meg3敲除模型中,发现胰岛素合成和分泌受损且Pdx1 和MafA 的表达降低,提示Meg3 可抑制β 细胞去分化[55]。miRNA 和lncRNA 在β细胞正常发育分化中发挥重要的作用,在T2D的发生发展中与β 细胞去分化的关系还需进一步的研究。
3.5 染色质修饰 染色质修饰包括DNA 甲基化、组蛋白修饰等。在T2D 的发生发展中,染色质修饰可影响β 细胞特性的维持。含SET 结构域的组蛋白甲基转移酶7/9(SET domain containing 7/9, SET7/9)参与组蛋白甲基化修饰。Mirmira 等[56]发现,Set7/9使Pdx1发生甲基化并增强其转录活性,在小鼠β 细胞中特异性敲除SET7/9后,Pdx1 表达水平下调,小鼠葡萄糖耐量和GSIS 功能受损。一项胰岛全基因组测序研究发现,T2D 患者胰岛的甲基化水平增加,且与对照者相比有25 820 个甲基化差异的区域(如Pdx1),提示胰岛细胞基因组DNA 甲基化失调可能与T2D发生发展相关[57]。
综上,β细胞去分化的机制与氧化应激、炎症、内质网应激、miRNA、lncRNA 和染色质修饰等相关。深入研究β 细胞去分化的关键环节和关键因子,可实现干预β 细胞的去分化,重建β 细胞功能为2 型糖尿病的防治提供新思路(图2)。
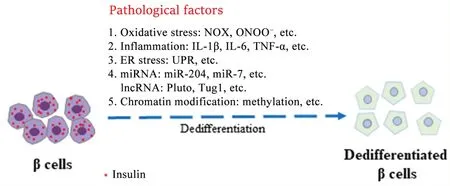
Figure 2. Schematic diagram of mechanisms of islet β cell dediffetentiation. NOX: NADPH oxidase; ONOO−: peroxynitrite; IL: in‐terleukin; TNF-α: tumor necrosis factor-α; ER: endoplasmic reticulum; UPR: unfolded protein response; miRNA: mi‐croRNA; lncRNA: long noncoding RNA; Pluto: Pdx1 locus upstream transcript; Tug1: taurine up-regulated gene 1.图2 胰岛β细胞去分化机制示意图
4 β细胞去分化的干预策略
现有的研究已证实,在去除高血糖和高脂血症等代谢刺激后β 细胞特征性的转录因子如Pdx1、Ma‐fA 和Nkx6.1 表达上调;同时如Aldh1a3、Ngn3、Sox9、NeuroD1和Oct4等胰腺祖细胞标志物表达下调,β 细胞分泌胰岛素的功能恢复,提示β 细胞去分化存在可逆性[58]。
有研究表明,减重、长期的饮食限制以及外科减肥手术等都可通过延缓β 细胞的去分化进而减轻T2D 患者的病情[25,31]。在干预β 细胞去分化方面,Son 等[59]发现,给db/db小鼠、饮食诱导的糖尿病小鼠或T2D 患者胰岛应用Aldh1a3 选择性抑制剂Kotx1,可以增加β 细胞胰岛素分泌并改善葡萄糖耐量,逆转β 细胞的去分化,这表明Aldh1a3 可能成为治疗T2D 的潜在治疗靶点。Zhang 等[60]发现,可以通过补充抗氧化剂谷胱甘肽逆转β 细胞去分化和衰竭,使已经下调的β 细胞特征性的转录因子包括Pdx1 和MafA 恢复表达,谷胱甘肽作为抗氧化剂可以预防长期血糖波动诱导的β 细胞胰岛素分泌功能障碍。目前在啮齿类动物和T2D 患者的研究中表明,通过缓解代谢刺激(如降低血糖或血脂水平)或干预β 细胞去分化可以延缓T2D 的发生发展或逆转T2D 模型鼠的疾病状态。未来需要进一步研究干预β 细胞去分化的措施,如减轻内质网应激、氧化应激和炎症等,延缓或逆转T2D中β细胞的去分化和功能丧失。
5 展望
胰岛β 细胞去分化导致的β 细胞功能障碍是T2D 发生发展的重要病理生理基础。深入阐明β 细胞去分化的原因及其病理机制,寻找延缓或逆转β细胞去分化的干预靶点及有效方法,是当前T2D 转化研究领域的前沿热点,其突破性进展可望对糖尿病的防治产生积极影响。
