腓骨肌萎缩症2家系临床及遗传学分析
2024-03-11李文武武豫冬梁继红董子梅文雪欢杨梦诗杨芳张婷孙浩
李文武,武豫冬,梁继红,董子梅,文雪欢,杨梦诗,杨芳,张婷,孙浩
腓骨肌萎缩症(CMT)是一组以进行性外周运动及感觉神经病变为特征的遗传性神经病[1-2],又称遗传性运动感觉神经病(HMSN),群体发病率为9.7/100000~82/100000[3]。传统分类可根据周围神经病变的类型[基于神经传导速度(NCV)]及遗传模式将CMT分为脱髓鞘型(CMT1,NCV<35 m/s)、轴索型(CMT2,NCV>45 m/s)及中间型(DI-CMT,NCV 35~45 m/s)[3]。由于基因检测技术在临床辅助诊断领域广泛应用,基于致病基因种类的新型分类系统,可以对上述3种CMT类型进行细分,如CMT1X(1型CMT,对应基因GJB1)等。当前人类在线孟德尔遗传数据库(OMIM)数据库收录的与CMT疾病明确相关基因已达62种(OMIM:PS118220)[4-9],这些基因在维持外周轴突、髓鞘、细胞信号和线粒体功能的结构完整性和功能完整性方面发挥了重要作用,如参与构成神经丝(NFs)的3种相关蛋白基因——神经丝轻链蛋白基因(NEFL)、神经丝中链蛋白基因(NEFM)及神经丝重链蛋白基因(NEFH),以及在线粒体融合及分裂状态之间维持动态平衡的MFN2基因。该4种基因对于神经元轴突的生长、轴突口径的维持和电信号沿轴突的传输均具有不可或缺的作用,其异常表达均与神经系统疾病相关。本研究对1例罕见CMT2 CC型(CMT2CC)疾病家系及1例CMT2 A2A型(CMT2A2A)家系进行临床及遗传学分析。其中,CMT2A2A家系携带MFN2基因NM_014874:c.746 C>G:p.S249C变异,该变异为首次报道,对MFN2致病性突变图谱进行了补充。在本文报道的CMT2CC疾病家系中,先证者携带NEFH基因NM_02107:c. 3057dupG:p.K1020Efs*43,该变异见于Ikenberg等[10]及Bian等[11]的发现。本研究为c.3057dupG变异致病性评价提供了更多临床证据。
1 临床资料
1.1 先证者
1.1.1 先证者1 患者,女性,33岁,因“进行性双下肢乏力三年余,加重半年余”于2021年5月20日入院。患者3年前出现进行性双下肢乏力,主要表现为上下楼梯费力及下蹲站立困难。入院查体:双下肢肌力Ⅲ+级,肌张力正常,大腿及臀部肌肉轻度萎缩,双下肢腱反射消失,弓形足,余未见明显异常。实验室检查:肌酸激酶292 U/L(参考范围40~200 U/L)。双侧大腿肌肉MRI检查:双侧大腿前肌群弥漫性萎缩(图1)。EMG显示上下肢周围神经源性损害(轴索为主)。对先证者1的基因组DNA进行全外显子捕获测序(WES)检测(武汉康盛达医学检验所),WES分析表型关键词汇为肌无力、高弓足、肌萎缩。结果显示,先证者1携带NEFH基因NM_021076:c.3057dupG:p.Lys1020Glufs*43杂合变异。该变异依据美国医学遗传学与基因组学学会(ACMG)指南[12]评级为可能致病(Likely Pathogenic:PS3_mode-rate+PM4+PM2+PP1)。此外,WES分析亦提示受检者携带表型相关但临床意义不明的变异位点DAG1:NM_004393.6:exon2:c.220G>A:p.V74I。DAG1基因对应疾病为抗肌萎缩相关糖蛋白性肌营养不良A9型(先天性伴脑和眼畸形)及抗肌萎缩相关糖蛋白性肌营养不良(肢带)C9型,二者均为常染色体隐性遗传,WES分析并未发现另一个构成符合杂合突变的致病位点。同时,两种对应疾病发病年龄均在新生儿期,与本案例家系疾病主诉不符。因此,排除该位点为先证者致病因素可能性。诊断为CMT2CC型,给予对症支持及康复治疗,目前患者病情稳定。
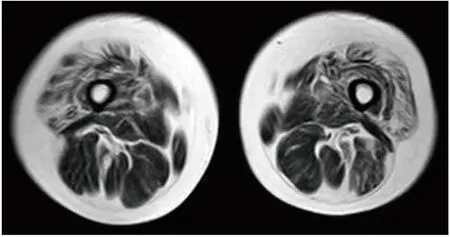
图1 先证者1肌肉MRI检查 双侧大腿T1WI示前肌群(股直肌、股中间肌、股内侧肌、股外侧肌、缝匠肌)弥漫性萎缩。
1.1.2 先证者2 患者,男性,6岁8个月。因“进行性行走困难伴频繁跌倒一年余”于2021年12月8日入院。家属发现患儿一年前行走时左右摆动,并进行性出现行走困难伴频繁跌倒。入院查体:神清语利,头颅五官正常,心肺腹未见异常,脊柱向右侧弯(图2),小腿中下1/3肌肉萎缩,四肢肌力及肌张力正常,双下肢腱反射消失。头颅MRI、心脏彩色多普勒检查正常。对先证者2的基因组DNA进行WES检测(武汉康盛达医学检验所),表型关键词汇为扁平足、脊柱侧弯、下肢肌萎缩。结果显示,先证者2携带MFN2基因NM_014874:c.746C>G:p.S249C杂合变异,依据ACMG规制评级为可能致病(Likely Pathogenic:PM1+PM2+PM5+PP3)。未见其他与表型相关的疑似致病性变异。诊断为CMT2A2A型,给予对症支持及康复治疗,目前患儿病情稳定。
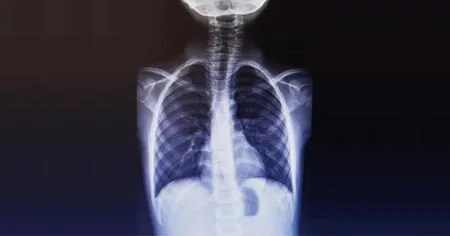
图2 先证者2胸部DR检查 胸片示脊柱向右侧弯。
1.2 家系调查
1.2.1 家系1 家系图见图3。先证者父亲、二姑及弟弟分别在40岁、41岁、29岁时出现相似症状,血肌酸激酶均增高,EMG提示上下肢周围神经源性损害(轴索为主)。先证者大姑已经去世,经先证者父亲口述大姑38岁时出现相似症状,双下肢乏力,加重伴行走不能两年余。先证者母亲无明显临床表型。一代验证结果显示(图4):先证者的父亲、二姑及弟弟均携带NEFH基因NM_021076:c.3057dupG:p.
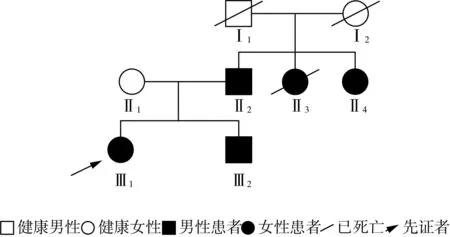
图3 家系1 CMT2CC型家系系谱图
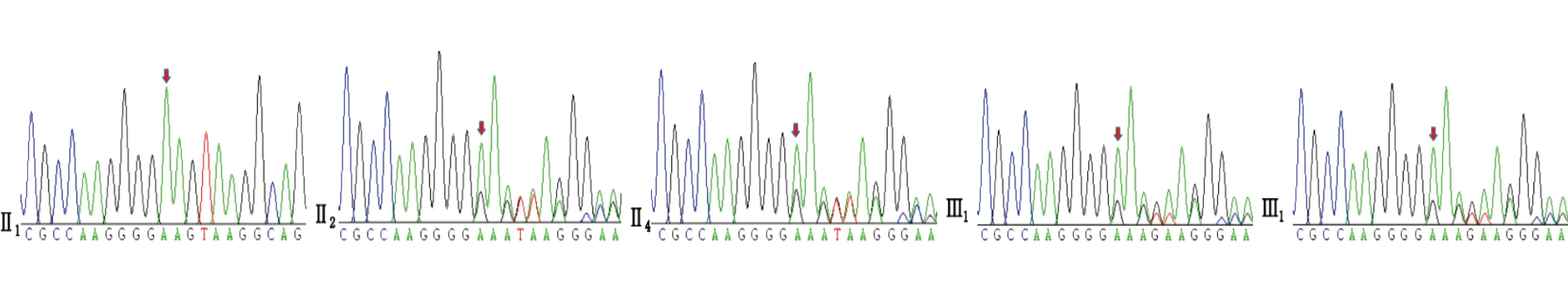
图4 家系1 NEFH基因NM_021076:c.3057dupG:p.Lys1020Glufs*43一代验证结果
Lys1020Glufs*43杂合变异,而临床未见异常的母亲未携带该变异。
1.2.2 家系2 家系图见图5。先证者父亲自幼步态异常,扁平足,鸭步;血肌酸激酶68 U/L,EMG检查示神经源损害,胸片正常。先证者母亲及姐姐临床表型未见异常。一代验证结果显示(图6),父亲同样携带MFN2基因NM_014874:c.746C>G:p.S249C杂合变异,而母亲及姐姐并未携带。
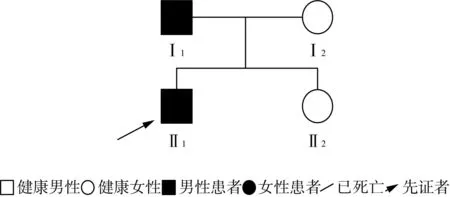
图5 家系2 CMT2A2A型家系系谱图
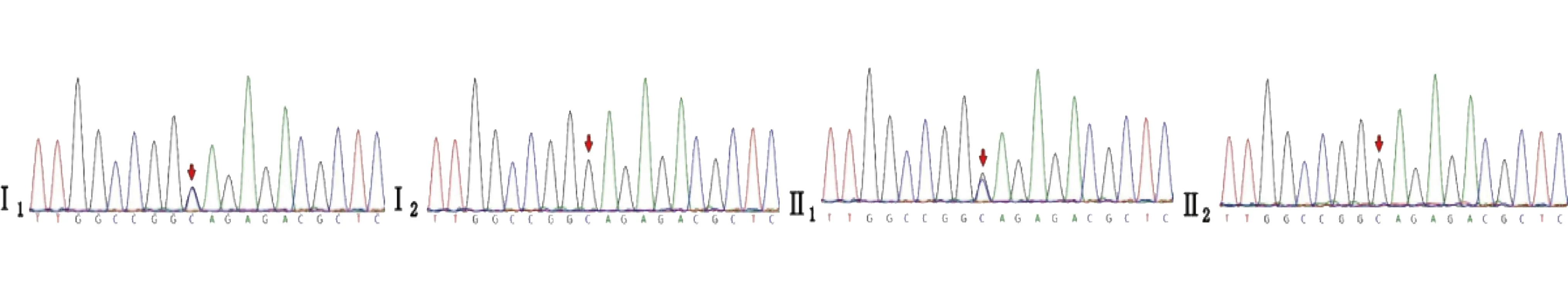
图6 家系2 MFN2基因NM_014874:c.746C>G:p.S249C一代验证结果
2 讨 论
CMT常在20岁之前发病,主要临床表现为远端肌无力、肌肉萎缩、腱反射减弱或消失、进行性对称性远端轻中度感觉减退,CMT临床进展缓慢,一般不会影响预期寿命[4]。本文报道了2个CMT2型腓骨肌萎缩症家系:CMT2CC(NEFH基因)及CMT2A2A(MFN2基因)。这两种类型均可出现远端肌肉萎缩、足畸形、NCV正常或轻度降低等临床表型,但因涉及基因不同,患者受累的器官及表型可存在自身的特点。2016年Rebelo等[13]首先报道CMT2CC,其呈常染色体显性遗传,由NEFH基因的致病性变异所致。目前在人类基因突变数据库(HGMD)数据库中收录的疾病致病性突变(DM)位点仅15处,且均位于NEFH基因4号外显子。无论是国外或是国内,该型CMT报道极其罕见,该疾病同其他类型CMT均起因于周围神经病变。临床特征包括进行性肌肉无力和肌肉萎缩,并主要影响下肢,常伴有轻度感觉缺陷和深部肌腱反射缺失。电生理学检测可见对称性的远端和近端感觉运动轴突神经病。除上述典型的CMT特征外,患者可有血清肌酸激酶增加(如本研究中家系1中受累成员)以及可能提示肌源性病变的EMG变化[10]。大部分患者在疾病的早期首先出现近端肌萎缩,而非远端[14],并且部分患者的临床表型可能更类似于脊髓性肌萎缩(SMA),而不是经典的CMT。本组家系1先证者携带NEFH基因NM_021076:c.3057dupG:p.Lys1020Glufs*43杂合变异。该变异可导致原有终止密码子丧失,造成蛋白编码序列的延长(PM4)。家系中患者表型与CMT2CC疾病表型相符,且先证者弟弟、父亲、二姑存在表型与变异共分离(PP1)。该变异在基因组聚合数据库(gnomAD)东亚人群中并无收录(PM2)。同时,功能研究提示c.3057dupG可造成NEFH蛋白产物在细胞内的异常集聚(PS3_moderate)[10]。依据ACMG对变异位点解释的规则,对c.3057dupG变异评级为可能致病。
CMT2A2A对应基因MFN2,为常染色显性遗传,常于儿童期起病。该型CMT为CMT2型分类中较为常见的一种[15]。HGMD数据库中对该基因收录的、已报道的DM位点达212处。其临床异质性较大,特征表现为患者下肢肌肉萎缩较上肢严重,并随着周围神经病变的进展而不断加重。上肢病变常从远端开始受累,运动障碍较感觉障碍明显。神经传导速度正常(NCV>42 m/s)或仅略有下降,体位性震颤常见。部分患者可出现听力损失、视神经萎缩,早发患者出现脊柱侧弯及认知衰退[16-17]。本组家系2先证者表型与CMT2A2A临床特征相符。基因检测显示,先证者携带MFN2基因NM_014874:c.746C>G:p.S249C杂合变异,一代验证提示变异来自父亲。尽管其父亲并无明显肌萎缩症状,但步态异常、扁平足等提示神经-肌肉病理改变表型。c.746C>G变异经查询HGMD、ClinVar等相关专业数据库,目前未见相关文献报道及收录。该变异发生在MFN2基因关键功能结构域Dynamin-type guanine nucleotide-binding (G) domain,且该区域缺少良性变异(PM1)。该位点在gnomAD数据库的东亚人群中无收录(PM2)。软件预测SIFT、Polyphen2、MutationTaster均提示有害(PP3)。有研究[18-19]报道,在同一位置其他类型碱基替换为致病性变异(NM_014874:c.746C>T: p.S249F)(PM5)。依据ACMG对变异位点解释的规则,对c.746C>G变异评级为可能致病。
目前,本病除了理疗、定制矫形支架或外科手术外,临床尚缺乏有效的治疗手段,药物治疗亦常用于神经性疼痛的缓解。本组病例均以对症支持治疗及康复训练为主,病情稳定。
本次研究进一步支持了NEFH基因NM_021076:c.3057dupG:p.Lys1020Glufs*43变异位点的致病性,为CMT2CC的临床诊断补充更多表型信息。MFN2基因NM_014874:c.746C>G:p.S249C变异可能为CMT2A2A新的致病位点。基因检测可为表型无特异性指证的神经肌肉疾病提供鉴别诊断依据。同时,更多的病例报道也为变异位点的致病性评级及解释提供有力的证据支持。
作者贡献说明李文武提出选题,设计、实施研究,采集、分析、解释数据,撰写、修改文章;武豫冬采集、统计分析数据,指导文章修改;梁继红、董子梅采集、统计分析数据,指导文章修改;文雪欢、杨梦诗指导文章修改、研究设计;孙浩指导选题、设计研究,指导起草、修改文章,提供研究经费、技术材料;杨芳、张婷指导文章修改,提供技术材料
