两种珍稀白蝶兰属(兰科)叶绿体基因组比较分析
2024-03-10汪雨唐露邵士成马长乐李健罗艳
汪雨 唐露 邵士成 马长乐 李健 罗艳


摘 要: 為理解珍稀濒危兰科植物龙头兰(Pecteilis susannae)和景洪白蝶兰(P. hawkesiana)的叶绿体基因组的基本特征,开发用于物种鉴定、保护遗传学和系统发育分析的分子标记,该研究利用二代测序技术对龙头兰和景洪白蝶兰进行浅层基因组测序,采用生物信息学分析方法进行叶绿体基因组的拼接、组装和注释,并与其他近缘物种进行比较基因组分析和系统发育分析。结果表明:(1)龙头兰和景洪白蝶兰的叶绿体基因组大小分别为154 407 bp和153 891 bp,由一对26 550 bp和26 523 bp的反向重复序列(IR)、84 204 bp和83 756 bp的大单拷贝区(LSC)、17 103 bp和17 089 bp的小单拷贝区(SSC)组成;均注释了111个唯一基因,包括77个蛋白质编码基因、30个tRNA基因和4个rRNA基因。(2)在叶绿体基因组中分别鉴定出94个和92个简单重复序列(SSRs)。 (3)二者之间存在706个单核苷酸多态性(SNPs)位点和152个插入缺失(InDels)位点,其中cpInDel 067等可以区分2个物种。(4)观察到1个差异较大的基因(accD)和9个高变区(rps19-psbA、matK-trnQ-UUG、psbM-psbD、trnT-UGU-ndhJ、accD-psaI、ycf4-cemA、clpP-psbB、ndhF-trnL-UAG、rps15-ycf1)。(5)系统发育分析结果显示,龙头兰、景洪白蝶兰和鹅毛玉凤花(Habenaria dentata)的亲缘关系较近。在白蝶兰属2种叶绿体基因组研究中获得的SSR位点、InDels和高变区序列可为物种鉴定、开发利用及其资源保护提供有价值的遗传信息。
关键词: 龙头兰, 景洪白蝶兰, 叶绿体基因组, 分子标记, 系统发育
中图分类号: Q943
文献标识码: A
文章编号: 1000-3142(2024)01-0043-13
Comparative analysis of chloroplast genomes of two
rare Pecteilis species (Orchidaceae)
WANG Yu1,2, TANG Lu1, SHAO Shicheng1, MA Changle2,3, LI Jian4, LUO Yan1*
( 1. Centre for Gardening and Horticulture, Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences, Mengla 666300, Yunnan,
China; 2. School of Landscape Architecture, Southwest Forestry University, Kunming 650224, China; 3. Southwest Landscape Architecture
Engineering Technology Research Center, State Forestry and Grassland Administration, Kunming 650224, China; 4. Orchid
Conservation & Research Center of Shenzhen and the National Orchid Conservation Center of China, Shenzhen Key
Laboratory for Orchid Conservation and Utilization, Key
Laboratory of National Forestry and Grassland
Administration for Orchid Conservation and
Utilization, Shenzhen 518114, Guangdong, China )
Abstract: Pecteilis susannae and P. hawkesiana are rare and endangered species with important medicine and ornament value. However, little is known about the genetic information of these two species. In order to understand the basic characteristics of the chloroplast genome of these two Pecteilis species, and to develop molecular markers for species identification, conservation genetic and phylogenetic analysis, the genome skimming approach using next-generation sequencing methods was used to generate chloroplast DNA sequences in this study. The chloroplast genomes were assembled and annotated by bioinformatics analysis. Simple sequence repeats (SSRs), single nucleotide polymorphisms (SNPs), and insertions and deletions (InDels) were identified. Furthermore, comparative chloroplast genomic and phylogenetic analyses were conducted with closely related species. The results were as follows: (1) The newly sequenced chloroplast genomes of P. susannae and P. hawkesiana were 154 407 bp and 153 891 bp in size. They comprised a pair of 26 550 bp and 26 523 bp inverted repeats (IR) that separated a large 84 204 bp and 83 756 bp single copy region (LSC) and a small 17 103 bp and 17 089 bp single copy region (SSC), respectively. Both chloroplast genomes contained 111 unique genes, including 77 protein-coding genes, 30 tRNA and 4 rRNA genes. (2) Ninety-four simple sequence repeats (SSRs) were identified in the P. susannae chloroplast genome and 92 in that of P. hawkesiana. (3) Comparisons of two chloroplast genomes revealed that there were nucleotide variations including 706 single-nucleotide polymorphism sites and 152 InDels between the two Pecteilis species, of which several markers (cpInDel 067) could discriminate the two Pecteilis species. (4) The one most divergent gene (accD) and the nine most divergent intergenic regions (rps19-psbA, matK-trnQ-UUG, psbM-psbD, trnT-UGU-ndhJ, accD-psaI, ycf4-cemA, clpP-psbB, ndhF-trnL-UAG, rps15-ycf1) among genomes were detected. (5) The phylogenetic analysis based on the chloroplast genome sequences revealed that P. susannae, P. hawkesiana and Habenaria dentata are closely related. The molecular markers (SSRs, InDels and hotspots) developed from the chloroplast genomes of two Pecteilis species in the present study can be used to identify related species and provide valuable genetic resources in utilizing and conserving natural resources.
Key words: Pecteilis susannae, P. hawkesiana, chloroplast genome, molecular markers, phylogeny
白蝶兰属(Pecteilis)为地生兰,隶属于兰科(Orchidaceae)红门兰亚科(subfamily Orchidoideae)红门兰族(tribe Orchideae)红门兰亚族(subtribe Orchidinae),全世界约有10种,主要分布于亚洲热带至亚热带地区(Jin et al.,2014,2017;Teoh,2021)。长期以来,白蝶兰属与红门兰亚族的玉凤花属(Habenaria)系统发育关系不清(Pridgeon et al.,2001)。2个属在形态学方面的关键区别在于合蕊柱的结构:白蝶兰属的花药室具宽的药隔,柱头无柄附着在唇瓣基部;玉凤花属的花药室的药隔较窄,柱头与唇瓣基部之间形成柱头枝(stigmaphore)(Wah et al.,2021)。此外,白蝶兰属的花通常比玉凤花属的大,唇瓣通常有较长的距。但是,根据最近的分子系统发育分析,白蝶兰属并非单系,嵌于玉凤花属中(Jin et al.,2014,2017)。
龙头兰(P. susannae)是白蝶兰属的模式种,也称白蝶花,从东喜马拉雅一直到东南亚和马来半岛都有分布(Wah et al.,2021)。在我国,龙头兰广泛分布于南部和西南部各省区,生长于海拔500~2 500 m的山坡林下开阔地、沟边和草坡(Wu et al.,2009)。虽然龙头兰在我国分布范围广,但近年来,生境丧失和过度采挖导致野生种群数量急剧下降。我国分布的白蝶兰属植物还包括滇南白蝶兰(P. henryi)、狭叶白蝶兰(P. radiate)和景洪白蝶兰(P. hawkesiana)。景洪白蝶兰分布于东南亚热带地区,2015年才在我国云南西双版纳自治州发现有分布,个体数量极少,非常稀有(Li et al.,2015)。目前,我国白蝶兰属植物的野生资源的基础调查尚不深入,遗传信息缺乏,极大地阻碍了对该属野生资源的保护和利用。
高等植物的叶绿体基因组(chloroplast genome,cpDNA)较小,通常为典型的环状四分体结构,由大单拷贝区(large single copy,LSC)、小单拷贝区(small single copy,SSC)和2个反向重复区(inverted repeats,IRs)构成(Ruhlman & Jansen,2014)。目前叶绿体基因组通过高通量的浅层基因组测序(genome skimming)技术较易获得(Fu et al.,2022; 黎若竹等,2022)。植物的叶绿体基因组较核基因组更具有保守性和遗传稳定性,进化速率适中,不存在基因重组现象等特征,在植物系统发育研究中得到广泛应用。叶绿体基因组序列揭示了许多基因结构变异,包括简单重复序列(simple sequence repeats,SSRs)、单核苷酸多态性(single nucleotide polymorphisms,SNPs)和插入缺失(insertions and deletions,InDels)等。齐丹等(2018)利用南方梨属(Pyrus)叶绿体基因组的6个片段发现秦岭淮河以南地区的砂梨和白梨亲缘关系较近,湖南地区的砂梨遗传多样性更丰富。汤晨茜等(2022)比较了陕甘花楸(Sorbus koehneana)和爪瓣花楸(S. unguiculata)的叶绿体基因组,探究二者的系统发育关系。陈模舜和杨仲毅(2022)利用26个天台鹅耳枥(Carpinus tientaiensis)叶绿体基因組的SNP进行分析,揭示天台鹅耳枥的遗传多样性和谱系分化。植物叶绿体基因组序列经常被用作DNA条形码(DNA barcoding)的分子标记进行物种鉴别,包括matK、rbcL、psbA-trnH和atpF-atpH等(Kress & Erickson,2007;Lahaye et al.,2008)。李镇兵等(2022)对3个品种木芙蓉(Hibiscus mutabilis)的叶绿体基因组进行分析后认为,使用ycf1、ndhB等基因可以对木芙蓉品种间及近缘种间进行鉴定。李冉郡等(2022)对大黄(Rheum spp.)药材基原物种的叶绿体基因组高变区进行特异DNA条形码开发,可以精准地鉴别3种大黄。姚辉等(2015)认为石斛属(Dendrobium)的psbK-psbI片段可以作为石斛属的候选分子标记,并成功利用其完成6份样品的鉴定。潘佳佳(2017)对高变位点进行筛选后,成功找到霍山石斛(D. huoshanense)的特异性位点,可以高效地将霍山石斛从各种枫斗类石斛产品中分辨出来。
本研究基于二代测序技术进行基因组浅层测序,利用生物信息学软件组装了龙头兰和景洪白蝶兰的叶绿体基因组,详细比较了2种白蝶兰属叶绿体基因组的结构差异,并与亲缘较近的玉凤花属植物进行比较和系统发育关系的分析,拟探讨:(1)龙头兰和景洪白蝶兰中的叶绿体基因组中有哪些位点可以作为特征性分子标记;(2)叶绿体基因组是否能够辨析白蝶兰属和玉凤花属的系统发育关系。
1 材料与方法
1.1 试验材料
龙头兰和景洪白蝶兰(图1)均保存于中国科学院西双版纳热带植物园保育苗圃中(101°46′ E、21°54′ N)。采集2种植物的新鲜叶片放置于硅胶中干燥保存。
1.2 DNA的提取和测序
取干燥后的龙头兰和景洪白蝶兰叶片,放入组织研磨器中充分研磨,按照植物总DNA提取试剂盒Tiangen DNA试剂盒(TIANGEN,中国)说明书使用方法进行叶片DNA的提取,并按照Illumina TruSeq文库制备试剂盒(Illumina,USA)构建DNA文库,测序文库由上海派森诺生物科技有限公司通过Illumina HiSeq 2500平台进行浅层基因组测序,测序读长为PE150。
1.3 叶绿体基因组的组装和注释
测序所得的原始数据经fastp软件(Chen et al.,2018)进行过滤,获得高质量的HQ data,之后使用GetOrganelle平台进行叶绿体基因组的组装(Jin et al.,2020),组装后获得的数据导入Bandage v0.8.1检查(Wick et al.,2015),判断其是否为双链环状四分体结构。确认组装所得的数据合格可用后,将其分别上传至GeSeq(Michael et al.,2017)和CPGAVAS2(Shi et al.,2019)平台进行叶绿体基因组注释。在NCBI(National Center for Biotechnology Information)数据库中检索兰科红门兰亚族已发表且注释的狭叶白蝶兰(KX871237)、线叶十字兰(H. linearifolia, NC_059696)和H. cruciformis(NC_059695)作为参考序列,将注释后获得的两组数据使用Geneious prime 2022.0.2软件进行人工校正比对。利用Organellar Genome DRAW(Greiner et al.,2019)在线工具绘制龙头兰和景洪白蝶兰的叶绿体基因组图谱。基因组长度、LSC区长度、SSC区长度、IR区长度、GC含量等通过Geneious prime 2022.0.2进行统计。
1.4 简单重复序列分析
利用MISA程序(Beier et al.,2017)预测龙头兰和景洪白蝶兰的简单重复序列(SSRs),单核苷酸的最小重复单位设置为10个,二核苷酸的最小重复单位设置为5个,三核苷酸的最小重复单位设置为4个,四核苷酸、五核苷酸和六核苷酸的最小重复单位设置为3个。
1.5 序列差异分析
使用Geneious prime 2022.0.2的插件MAFFT Alignment将2种白蝶兰属的叶绿体基因组序列进行比对,通过BioEdit软件编辑整理,使用DnaSP v6.12.03计算单核苷酸变异(SNPs)和插入缺失(InDels)的数量,每100 bp计算突变率,并评估2种白蝶兰属植物的叶绿体基因组的核苷酸多样性(Pi)值,当Pi值高于0.030 0时,将其定义为高变区。
1.6 比较基因组分析
从NCBI上下载已公布的1种白蝶兰属和3种玉凤花属的叶绿体基因组,包括狭叶白蝶兰、鹅毛玉凤花(H. dentate,OK012095)、线叶十字兰、H. cruciformis,与本研究组装的龙头兰和景洪白蝶兰,利用mVISTA程序(Frazer et al.,2004))对3种白蝶兰属和3种玉凤花属的叶绿体基因组序列进行比较。通过R v4.1.3软件运行IRscope脚本(Amiryousefi et al.,2018),比较6个物种中LSC区、SSC区和IR区的边界基因。
1.7 系统发育分析
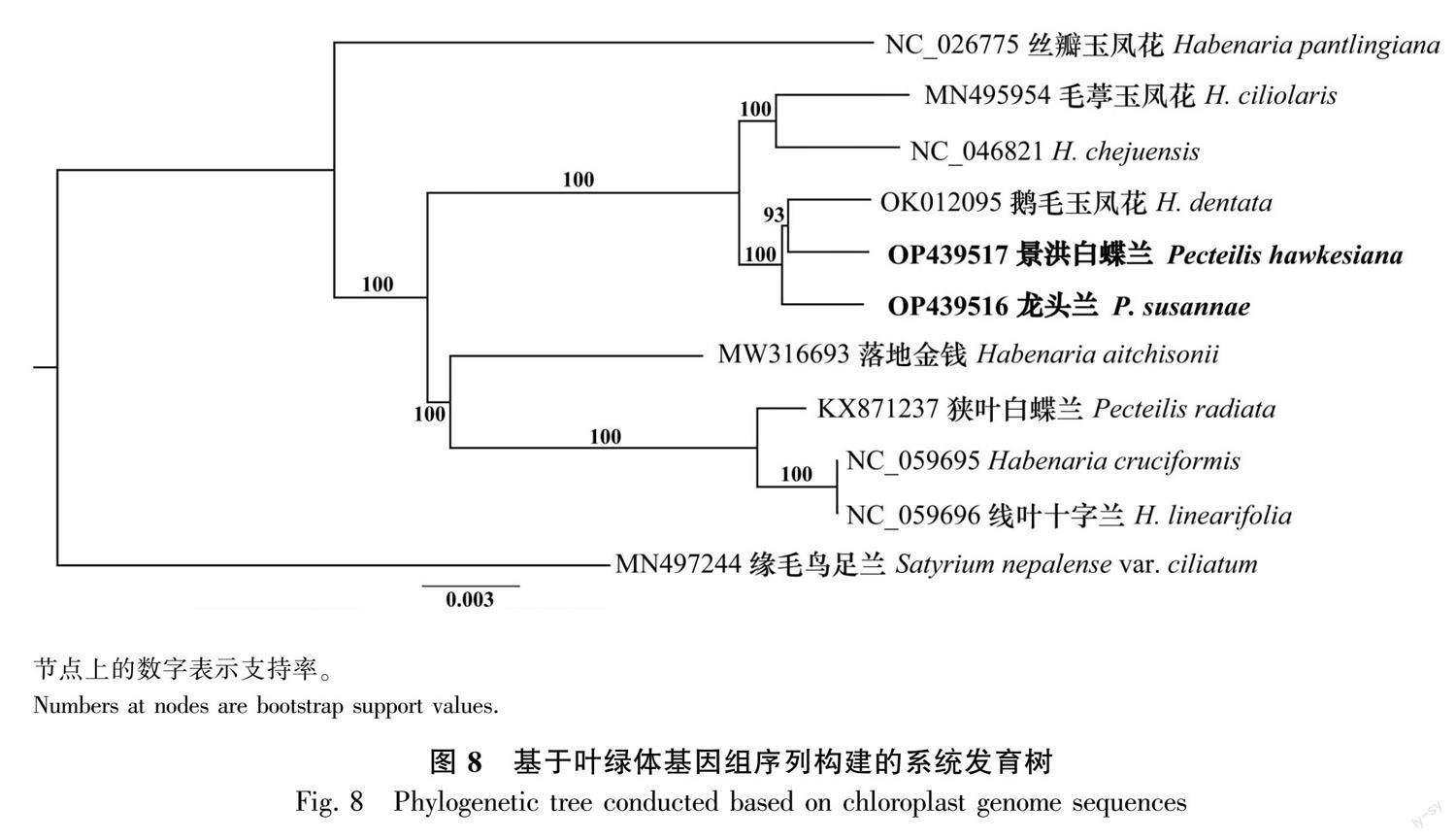
除了已下载的上述4种白蝶兰属和玉凤花属植物以外,再从NCBI上下载另外4种已公布的玉凤花属植物的叶绿体基因组序列,包括丝瓣玉凤花(H. fordii,NC_026775)、毛葶玉凤花(H. ciliolaris,MN495594)、H. chejuensis(NC_046821)、落地金钱(H. aitchisonii,MW316693),以红门兰亚族的缘毛鸟足兰(Satyrium nepalense var. ciliatum,MN497244)为外类群(Jin et al.,2017),将龙头兰和景洪白蝶兰的叶绿体基因组序列与这些近缘物种进行系统发育关系分析。通过PhyloSuite v1.2.2平台(Zhang et al.,2020)查找最适模型后使用IQ-TREE构建Maximum Likelihood(ML)系统发育树。通过FigTree v1.4.0软件编辑系统发育树。
2 结果与分析
2.1 龙头兰和景洪白蝶兰叶绿体基因组的基本特征
在去除低质量reads后,分别得到了龙头兰和景洪白蝶兰3.37 Gb和1.07 Gb的叶绿体基因组序列数据。在组装之后通过Bandage v0.8.1软件检查,判断组装后的数据可用。基因组结构为闭合的环状四分体结构,与典型的被子植物叶绿体基因组结构相同。
2个白蝶兰属植物的叶绿体基因组的基因图谱如图2所示。龙头兰叶绿体基因组全长154 407 bp,包含一对26 550 bp的IR区,由84 204 bp的LSC区和17 103 bp的SSC区分开。景洪白蝶兰叶绿体基因组全长153 891 bp,包含一对26 523 bp的IR区,由83 756 bp的LSC区和17 089 bp的SSC区分开(表1)。龙头兰叶绿体基因组的GC含量为36.5%,景洪白蝶兰的GC含量为36.6%,二者都是在IR区的GC含量最高(43.0%),并且高于基因组的GC含量。龙头兰和景洪白蝶兰的叶绿体基因组均注释了111个唯一基因(unique genes),包含77个蛋白质编码基因、30个转运RNA(tRNA)基因和4个核糖体RNA(rRNA)基因(表2)。其中,18个基因含有1个内含子,2个基因(pafI和clpP 1)含有2个内含子(表2)。在所有基因中,有19个基因位于反向重复区且具有2个拷贝(ndhB、rps7、rps12、rps19、rpl2、rpl23、rrn4.5、rrn5、rrn16、rrn23、trnA-UGC、trnH-GUG、trnI-CAU、trnI-GAU、trnL-CAA、ycf2、trnN-GUU、trnR-ACG和trnV-GAC,表2)。注釋的龙头兰和景洪白蝶兰的叶绿体基因组信息已提交至GenBank上,序列号分别为OP435916和OP435917。
2.2 叶绿体基因组中的简单重复序列特征
在龙头兰的叶绿体基因组中共检测到94个SSRs,包括4种类型的SSRs,单核苷酸、二核苷酸、三核苷酸和四核苷酸重复序列分别为74、16、1、3个,其中54个SSRs位于基因间隔区(intergenic spacer,IGS)、17个SSRs位于蛋白质编码区(coding sequence,CDS)、23个SSRs位于内含子上(图3)。在景洪白蝶兰的叶绿体基因组中共检测到92个SSRs,包括5种类型的SSRs,单核苷酸、二核苷酸、三核苷酸、四核苷酸和五核苷酸重复序列分别为72、15、1、3、1个,其中,58个SSRs位于IGS中,15个SSRs位于CDS中,19个SSRs位于内含子中(图3)。在单核苷酸SSRs中,龙头兰和景洪白蝶兰分别有3个和4个C/G型,其余均为A/T型。大多数二核苷酸SSRs为AT/TA型。这一结果与叶绿体基因组的SSRs通常由A/T组成而很少包含C/G的观点一致(Kuang et al.,2011;陈模舜和杨仲毅,2022)。
2.3 叶绿体基因组中的突变和插入缺失
比对2种白蝶兰属植物的叶绿体基因组序列,发现共有706个点突变。其中IR区、LSC区、SSC区发生点突变次数分别为48、517、141次(表3)。706个SNPs标记中包括259个转换和447个颠换(图4)。经统计发现,在两条叶绿体基因组序列中,发生突变频次最高的区域为内含子,每100 bp大约会发生6.238 53次突变事件。
在2种白蝶兰属植物的叶绿体基因组中共检测到InDels 152个,其中IR区、LSC区、SSC区分别产生了8、131、13个InDels(表3)。内含子区域每100 bp会发生1.743 12次InDels事件。而蛋白质编码区相对保守,每100 bp仅发生0.013 94次。在LSC区trnL-UAA和trnF-GAA 2个基因之间的序列中发现了最长的InDels(104 bp)。另外3个较长的InDels也在LSC区,分别位于trnT-GGU和psbD基因之间(84 bp)、ndhC和trnV-UAC基因之间(54 bp)、trnL-UAA基因内(50 bp)。这4个较大的InDels可以作为潜在的分子标记开发区域,用来特异性识别龙头兰和景洪白蝶兰。
2.4 叶绿体基因组序列的核苷酸多样性
通过计算龙头兰和景洪白蝶兰的叶绿体基因组间的核苷酸多样性(Pi)值可知,基因间隔区的Pi值为0~0.053 4(图5: A),编码区的Pi值为0~0.018 9(图5: B),表明二者的编码区序列相对保守。大多数的高变位点位于LSC区,其次是SSC区,IR区序列的Pi值较低。在非编码区中,petG-trnW-CCA的核苷酸多样性值最高(Pi=0.053 4),除此之外,还有5个基因间隔区rps16-trnQ-UUG、rps14-psaB、petD-rpoA、rpl16-rps3、rrn4.5-rrn5(Pi>0.030 0)可视为高变区。在编码区中,psaJ基因的核苷酸多样性值最高(Pi=0.018 9),以及其他2个基因trnS-GGA(Pi=0.011 5)和rpl32(Pi=0.011 5)的核苷酸多样性值均大于0.010 0,可视为多样性较高的基因。这些核苷酸多样性程度高的基因和基因间隔区可开发为潜在的物种鉴定的特异性分子标记。
2.5 白蝶兰属和玉凤花属的基因组比较
笔者将新测序的2种白蝶兰属植物与其近缘种狭叶白蝶兰、鹅毛玉凤花、线叶十字兰和H. cruciformis共6个种的叶绿体基因组的边界基因进行了比较分析。叶绿体基因组边界收缩扩张分析显示(图6),6个种在LSC/IRb、IRb/SSC、SSC/IRa和IRa/LSC边界都具有相同的基因,包括rpl22、ndhF、ycf1、rps19和psbA,并且收缩和扩张的程度较为相似。Rpl22基因跨LSC/IRb边界,6个种仅有1 bp之差。ndhF基因跨IRb/SSC边界,6个种仅有6 bp之差,其中龙头兰、景洪白蝶兰和鹅毛玉凤花相同,而狭叶白蝶兰、线叶十字兰和H. cruciformis相同。ycf1基因跨SSC/IRa边界,在龙头兰和鹅毛玉凤花中延伸至IR区993 bp,在景洪白蝶兰和狭叶白蝶兰中延伸至IR区1 002 bp,在线叶十字兰和H. cruciformis中延伸至IR区1 032 bp(图6)。从边界分析来看,白蝶兰属3种和玉凤花属3种之间无明显差异,表明叶绿体基因组结构不支持2个属的划分。
以注释的狭叶白蝶兰序列为参考序列,利用在线软件mVISTA分析了6个物种叶绿体基因组的序列差异(图7)。由图7可知,6个物种中LSC区和SSC区的差异性高于IR区,非编码区的差异性高于编码区。其中,差异性较大的基因为accD,差异性较高的区域有rps19-psbA、matK-trnQ-UUG、psbM-psbD、trnT-UGU-ndhJ、accD-psaI、ycf4-cemA、clpP-psbB、ndhF-trnL-UAG、rps15-ycf1等区域,可以利用这些区域,开发特异性标记,进行物种鉴定和系统发育研究。
2.6 系统发育分析
通过PhyloSuite获取最适模型为TVM+R2+F,采用此模型构建所选白蝶兰属和玉凤花属的系统发育树,以缘毛鸟足兰作为外类群。由图8可知,10种白蝶兰属和玉凤花属植物形成了3个分支,其中丝瓣玉凤花单独分出来为基部分支, 毛葶玉凤花、H. chejuensis、鹅毛玉凤花、景洪白蝶兰、龙头兰聚为一个单系分支,而落地金钱、狭叶白蝶兰、H. cruciformis、线叶十字兰聚为一个单系分支。龙头兰、鹅毛玉凤花和景洪白蝶兰则聚为一个亚分支,而后二者形成了姐妹群的關系,表明三者的亲缘关系较近。2种白蝶兰属嵌入玉凤花属,表明这2属从叶绿体基因组构建的系统发育树上不能分开。
3 讨论与结论
3.1 基因组序列特征比较分析
在兰科红门兰亚族中玉凤花属、舌唇兰属、鸟足兰属(Satyrium)、无柱兰属(Amitostigma)、手参属(Gymnadenia)等属已有叶绿体基因组的报道,基因组大小为146 754~156 120 bp(Kim et al.,2020)。本文首次报道并解析了红门亚兰族中白蝶兰属2种的叶绿体基因组的序列特征。龙头兰和景洪白蝶兰的叶绿体基因组结构与大多数被子植物叶绿体基因组结构相似, 都为典型的环状四分体结构,它们的长度、基因组结构、基因数目、GC含量等均与以前报道的红门兰族物种的基因组特征相似。已报道的H. cruciformis、狭叶白蝶兰等物种的叶绿体基因组均编码了113个唯一基因,包括79个蛋白质编码基因、30个tRNA和4个rRNA,GC含量为36.6%(Kim et al.,2017,2020);在本研究中,龙头兰和景洪白蝶兰都包含111个唯一基因,包括77个蛋白质编码基因、30个tRNA基因和4个rRNA基因,GC含量分别为36.5%和36.6%。在兰科植物叶绿体基因组中编码NAD(P)H脱氢酶复合体的ndh基因截短或丢失的现象较为多见,但在红门兰亚科中很少发生(Kim et al.,2020)。丝瓣玉凤花的叶绿体基因组中具有11个ndh基因(Lin et al.,2015),本研究中龙头兰和景洪白蝶兰的叶绿体基因组中也包含了完整基因结构的11个ndh基因,没有截短和丢失的现象。
3.2 特异分子鉴定标记的筛选
本研究通过基因组的比较研究,在2种白蝶兰属植物中鉴定出的SSRs、InDels和核苷酸序列高变区等均可作为特异的分子标记鉴定物种。本研究发现龙头兰叶绿体基因组中共有94个SSRs,景洪白蝶兰叶绿体基因组中共有92个SSRs,二者在SSRs数量上仅相差2个。龙头兰不存在五核苷酸重复序列,景洪白蝶兰有1個五核苷酸重复。已报道的同属狭叶白蝶兰中共检测到76个SSRs位点,包括58个单核苷酸SSRs、17个二核苷酸SSRs和1个三核苷酸SSRs(Kim et al.,2017)。这些SSRs位点可进一步开发为遗传标记用于遗传多样性研究及同属近缘种间鉴别的分子标记。InDel标记具有稳定性好、多态性高、分型系统简单等优点,在作物育种、医学诊断等领域多有应用(杨洁等,2016)。在黄麻(Corchorus capsularis)和长蒴黄麻(C. olitorius)中共鉴定出了294个InDels,其中cpIndel 205可以将这2种黄麻属植物区分开来(Fang et al.,2021)。本研究在龙头兰和景洪白蝶兰叶绿体基因组中共鉴定出152个InDels,其中cpInDel 067(104 bp)位于大单拷贝区trnL-UAA基因和trnF-GAA基因之间,全长超过100 bp,可以作为潜在的区分2个物种的分子标记。高变序列较短,作为DNA条形码可以经济、快速地区分同属近缘物种(杨嘉鹏等,2020)。Li等(2020)利用叶绿体基因组的LSC区可高效、精准地对枫斗类石斛进行中药材鉴定。姚辉等(2015)利用psbK-psbI片段可成功分辨18种药用石斛及其混伪品。杨嘉鹏等(2020)筛选出5个高度变异的基因间隔区序列(psbI-trnS、psbC-trnS、clpP-ex1-psbB、psaJ-rpl33、rpl33-rps18)可用于药用石豆兰(Bulbophyllum)的鉴定。本研究中2种白蝶兰属植物中的多数高变位点位于LSC区,基因间隔区序列petG-trnW-CCA、rps16-trnQ-UUG、rps14-psaB、petD-rpoA、rpl16-rps3、rrn4.5-rrn5为高变区,可用于中药材龙头兰及其混淆种的鉴别。
3.3 基于叶绿体基因组的白蝶兰属和玉凤花属的系统发育关系
长期以来,关于红门兰亚族内的进化与分类问题争议不断。在白蝶兰属与玉凤花属中具宽大扇形的唇瓣侧裂片的种类在形态上最为接近,传统的形态学分类主要是根据柱头是否有柄将二者区分开来(Wah et al.,2021)。但是,形态学特征往往变异丰富、个体差异较大。因此,我们需要结合更加充分的分子证据。玉凤花属和白蝶兰属的遗传背景资料尚十分缺乏,仅报道了为数不多的一些物种的叶绿体基因组。本研究通过GenBank检索到的玉凤花属和白蝶兰属物种的叶绿体基因组序列构建了基于叶绿体基因组的系统发育树,发现景洪白蝶兰和鹅毛玉凤花亲缘关系最近,并与龙头兰聚为一个单系分支,这一单系分支与Jin等(2014,2017)基于2个核基因和5个叶绿体基因片段得到的结果一致。龙头兰和景洪白蝶兰在花形态上与分布于亚洲热带的玉凤花属的一些类群非常接近,如花白色、花大、宽药隔、唇瓣具扇形裂片等。因此,二者的柱头是否有柄的形态特征不能作为分属的特征。Jin等(2017)指出柱头有柄的特征在这一单系分支中至少独立消失了2次。同属于白蝶兰属的分布于温带的狭叶白蝶兰并未与龙头兰和景洪白蝶兰聚成一支,而是和玉凤花属的H. cruciformis和线叶十字兰亲缘关系更近。Kim等(2017)和Tachibana等(2021)认为该种属于玉凤花属,本研究支持该观点,狭叶白蝶兰应置于玉凤花属中。本研究初步的系统发育结果与Jin等(2014,2017)基于核基因和叶绿体基因片段的分子系统发育分析得出的结果一致,均认为目前划分的白蝶兰属并非单系,与玉凤花属嵌套不能分开,可归并至玉凤花属中。完整的叶绿体基因组长度约15 kb,包含77~78个蛋白质编码基因,通过叶绿体基因组建树,序列长度、变异率和信息位点均大大增加,物种间关系的支持率都很高,多数分支的支持率高达100%,表明利用叶绿体基因组序列构建系统的发育树为解决白蝶兰属和玉凤花属物种之间的物种关系提供了有用信息,在今后的研究中,可在进一步全面取样的基础上,探讨这一复杂类群的种间系统发育关系。
致谢 中国科学院西双版纳热带植物园王晓静提供照片,谨致谢意。
参考文献:
AM IRYOUSEFI A, HYVNEN J, POCZAI P, 2018. IRscope: an online program to visualize the junction sites of chloroplast genomes[J]. Bioinformatics, 34(17): 3030-3031.
BEIER S, THIEL T, MUENCH T, et al., 2017. MISA-web: a web server for microsatellite prediction[J]. Bioinformatics, 33(16): 2583-2585.
CHEN MS, YANG ZY, 2022. Genealogical structure and differentiation analysis of Carpinus tientaiensis based on single nucleotide polymorphism of chloroplast genome[J]. Guihaia, 42(10): 1703-1716. [陳模舜, 杨仲毅, 2022. 基于叶绿体基因组SNP的天台鹅耳枥谱系结构与分化分析[J]. 广西植物, 42(10): 1703-1716.]
CHEN SF, ZHOU YQ, CHEN YR, et al., 2018. Fastp: an ultra-fast all-in-one FASTQ preprocessor[J]. Bioinformatics, 34(17): 884-890.
FANG SS, ZHANG LM, QI JM, et al., 2021. De novo assembly of chloroplast genomes of Corchorus capsularis and C. olitorius yields species-specific InDel markers[J]. Crop J, 9(1): 216-226.
FRAZER KA, PACHTER L, POLIAKOV A, et al., 2004. VISTA: computational tools for comparative genomics[J]. Nucleic Acids Res, 32(Suppl. 2): W273-W279.
FU CN, MO ZQ, YANG JB, et al., 2022. Testing genome skimming for species discrimination in the large and taxonomically difficult genus Rhododendron[J]. Mol Ecol Resour, 22(1): 404-414.
GREINER S, LEHWARK P, BOCK R, 2019. Organellar-GenomeDRAW(OGDRAW)version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes[J]. Nucleic Acids Res, 47(W1): W59-W64.
JIN JJ, YU WB, YANG JB, et al., 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes[J]. Genome Biol, 21(1): 241.
JIN WT, JIN XH, SCHUITEMAN A, et al., 2014. Molecular systematics of subtribe Orchidinae and Asian taxa of Habenariinae (Orchideae, Orchidaceae) based on plastid matK, rbcL and nuclear ITS[J]. Mol Phylogenet Evol, 77: 41-53.
JIN WT, SCHUITEMAN A, CHASE MW, et al., 2017. Phylogenetics of subtribe Orchidinae s.l. (Orchidaceae; Orchidoideae) based on seven markers (plastid matK, psaB, rbcL, trnL-F, trnH-psbA, and nuclear nrITS, Xdh): implications for generic delimitation[J]. BMC Plant Biol, 17(1): 222.
KIM C, DO HDK, JUNG J, et al., 2020. Characterization of the complete chloroplast genome of Korean endemic, Habenaria cruciformis (Orchidaceae)[J]. Mitochondrial DNA Part B, 5(3): 3287-3289.
KIM YK, JO S, CHOON SH, et al., 2020. Plastome evolution and phylogeny of Orchidaceae, with 24 new sequences[J]. Front Plant Sci, 11: 22.
KIM YK, KWAK MH, HONG JR, et al., 2017. The complete plastome sequence of the endangered orchid Habenaria radiata (Orchidaceae)[J]. Mitochondrial DNA Part B, 2(2): 704-706.
KRESS WJ, ERICKSON DL, 2007. A two-locus global DNA Barcode for land plants: the coding rbcL gene complements the non-coding trnH-psbA spacer region[J]. PLoS ONE, 2(6): e508.
KUANG DY, WU H, WANG YL, et al., 2011. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): implication for DNA barcoding and population genetics[J]. Genome, 54(8): 663-673.
LAHAYE R, VAN DER BANK M, BOGARIN D, et al., 2008. DNA barcoding the floras of biodiversity hotspots[J]. Proc Natl Acad Sci USA, 105(8): 2923-2928.
LI JW, YE DP, LIU JX, et al., 2015. Orchids newly recorded to China[J]. Plant Divers Resour, 37(3): 246-252.
LI LD, JIANG Y, LIU YY, et al., 2020. The large single-copy (LSC) region functions as a highly effective and efficient molecular marker for accurate authentication of medicinal Dendrobium species[J]. Acta Pharm Sin B, 10(10): 1989-2001.
LI RJ, WU LW, XIN TY, et al., 2022. Analysis of chloroplast genomes and development of specific DNA barcodes for identifying the original species of Rhei Radix et Rhizoma[J]. Acta Pharm Sin, 57(5): 1495-1505. [李冉郡, 武立偉, 辛天怡, 等, 2022. 大黄药材基原物种叶绿体基因组分析与特异DNA条形码开发[J]. 药学学报, 57(5): 1495-1505.]
LI RZ, CAI J, YANG JB, et al., 2022. Plastid phylogenomics resolving phylogenetic placement and genera phylogeny of Sterculioideae (Malvaceae s. l.)[J]. Guihaia, 42(1): 25-38. [黎若竹, 蔡杰, 杨俊波, 等, 2022. 利用叶绿体基因组数据解析锦葵科梧桐亚科的系统位置和属间关系[J]. 广西植物, 42(1): 25-38.]
LI ZB, REN T, DENG JJ, et al., 2022. Comparative analysis of the chloroplast genomes of three cultivars of Hibiscus mutabilis and its related species [J]. Guihaia, 42(12): 2007-2020. [李镇兵, 任婷, 邓姣姣, 等, 2022. 木芙蓉三个品种及近缘种的叶绿体基因组比较分析[J]. 广西植物, 42(12): 2007-2020.]
LIN CS, CHEN JJW, HUANG YT, et al., 2015. The location and translocation of ndh genes of chloroplast origin in the Orchidaceae family[J]. Sci Rep, 5(1): 1-10.
MICHAEL T, PASCAL L, TOMMASO P, et al., 2017. GeSeq-versatile and accurate annotation of organelle genomes[J]. Nucleic Acids Res, 45(W1): W6-W11.
PAN JJ, 2017. Analysis of genetic relationship of Dendrobium huoshanense population and its identification based on chloroplast genome[D]. Nanjing: Nanjing Normal University. [潘佳佳, 2017. 基于叶绿体基因组的霍山石斛居群亲缘关系分析及其鉴定研究[D]. 南京: 南京师范大学.]
PRIDGEON AM, CRIBB PJ, CHASE MW, et al., 2012. Genera Orchidacearum: Vol. 2, Orchidoideae (Part 1)[M]. Oxford: Oxford University Press.
QI D, CHANG YJ, CAO YF, et al., 2018. Genetic diversity and phylogenetics of pear(Pyrus L.)germplasm resources from south China revealed by chloroplast DNA[J]. Acta Hortic Sin, 45(12): 2308-2320. [齐丹, 常耀军, 曹玉芬, 等, 2018. 基于叶绿体DNA信息的南方梨属种质的遗传多样性和演化分析[J]. 园艺学报, 45(12): 2308-2320.]
RUHLMAN TA, JANSEN RK, 2014. The plastid genomes of flowering plants[J]. Meth Mol Biol, 1132: 3-38.
SHI LC, CHEN HM, JIANG M, et al., 2019. CPGAVAS2, an integrated plastome sequence annotator and analyzer[J]. Nucleic Acids Res, 47(W1): W65-W73.
TACHIBANA T, NISHIKAWA Y, KUBO N, et al., 2021. Morphological and genetic diversities of Habenaria radiata (Orchidaceae)in the Kinki area, Japan[J]. Int J Mol Sci, 22(1): 311.
TANG CQ, QIU ZX, TAN C, et al., 2022. Sorbus koehneana (Rosaceae): its complete chloroplast genome and phylogenetic relationship with S. unguiculata[J]. Acta Hortic Sin, 49(3): 641-654. [汤晨茜, 仇志欣, 檀超, 等, 2022. 陕甘花楸叶绿体基因组及其与爪瓣花楸的系统关系[J]. 园艺学报, 49(3): 641-654.]
TEOH ES, 2021. Pecteilis Rafin.[M]. Cham: Springer International Publishing: 231-233.
WAH LK, KUMAR P, GALE SW, 2021.971. Pecteilis susannae: Orchidaceae[J]. Curtiss Bot Mag, 38(1): 57-71.
WICK RR, SCHULTZ MB, ZOBEL J, et al., 2015. Bandage: interactive visualization of de novo genome assemblies[J]. Bioinformatics, 31(20): 3350-3352.
WU ZY, RAVEN PH, HONG DY, 2009. Flora of China: Vol. 25 (Orchidaceae)[M]. Beijing: Science Press; St. Louis: Missouri Botanical Garden Press: 136-137.
YANG J, HE J, WANG DB, et al., 2016. Progress in research and application of InDel markers[J]. Biodivers Sci, 24(2): 237-243.[楊洁, 赫佳, 王丹碧, 等, 2016. InDel标记的研究和应用进展[J]. 生物多样性, 24(2): 237-243.]
YANG JP, ZHU ZL, FAN YJ, et al., 2020. Comparative plastomic analysis of three Bulbophyllum medicinal plants and its significance in species identification[J]. Acta Pharm Sin, 55(11): 2736-2745. [杨嘉鹏, 朱紫乐, 范雅娟, 等, 2020. 三种石豆兰属药用植物的叶绿体基因组比较分析及其在物种鉴定中的意义[J]. 药学学报, 55(11): 2736-2745.]
YAO H, YANG P, ZHOU H, et al., 2015. Identification of medicinal plant Dendrobium based on the chloroplast psbK-psbI intergenic spacer[J]. Acta Pharm Sin, 50(6): 783-787. [姚辉, 杨培, 周红, 等, 2015. 基于叶绿体psbK-psbI序列的石斛属药用植物鉴定[J]. 药学学报, 50(6): 783-787.]
ZHANG D, GAO F, JAKOVLIC I, et al., 2020. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies[J]. Mol Ecol Resour, 20(1): 348-355.
( 责任编辑 邓斯丽 )
收稿日期: 2023-09-01
基金项目: 深圳市濒危兰科植物保护与利用重点实验室开放基金(OU202204); 中国科学院西双版纳热带植物园园林园艺中心研究基金(E2ZK291B05); 国家自然科学基金(32270225)。
第一作者: 汪雨(1997-),硕士研究生,研究方向为园林植物资源与应用,(E-mail)wangyu@swfu.edu.cn。
* 通信作者: 罗艳,博士,研究员,研究方向为兰科植物多样性与保护,(E-mail)luoyan@xtbg.org.cn。
