皮肤脱屑相关遗传病以及一种全新疾病的命名建议
2024-02-29巩卓青汪慧君林志淼
巩卓青,汪慧君,林志淼
[作者单位]南方医科大学皮肤病医院,广东 广州 510091
皮肤是人体和外界环境直接接触的器官,有重要的屏障作用。表皮最外层的角质层是第一道物理性屏障,保护人体不受外界有害物质或刺激的侵袭,同时可防止体内的水分、电解质等重要成分过度流失,维持皮肤稳态。人体皮肤角质层由10~20层角化细胞和细胞间结构蛋白、细胞间脂质、角质桥粒(corneodesmosome)以及各种微生物群组成,这些成分构成了角质层特征性的“砖-墙”结构。而仅存在于角质层中的角质桥粒,是角质细胞间最主要的连接结构,其正常形成和降解是皮肤脱屑的关键因素。
1 皮肤生理脱屑及其影响因素
1.1皮肤脱屑 皮肤脱屑是指角质细胞不断脱落这一生理过程,角质桥粒被逐渐水解是其关键因素。角质桥粒由颗粒层的细胞间连接结构桥粒转化而来,主要由桥粒芯蛋白1(desmoglein-1,DSG1)、桥粒糖蛋白1(desmocollin-1,DSC1)和角膜锁链蛋白(corneodesmosin,CDSN)等结构蛋白构成。其中,DSG1和DSC1,两种桥粒跨膜糖蛋白,也是桥粒的结构蛋白,在角质桥粒形成过程中保留并通过转谷氨酰胺酶1、3和5(TGM1、TGM3和TGM5)介导的广泛酶交联锚定在角质细胞外周;而CDSN是角质桥粒特有的结构蛋白,通过板层小体分泌到颗粒层最上层的细胞间隙,嵌入DSG1、DSC1中进一步增强其细胞间连接作用[1-2]。完整的角质桥粒主要存在于角质层下层的角质细胞间,随着角质细胞上移,DSG1、DSC1以及CDSN被逐步水解成小分子,导致角质桥粒被逐渐水解,角质层最外层的角质细胞有序脱落。
1.2皮肤脱屑的影响因素 皮肤脱屑过程受到多种因素的精细调控,包括蛋白酶、内源性蛋白酶抑制剂和环境等。激肽释放酶相关肽酶(kallikrein-related peptidase,KLK)是降解角质桥粒的主要蛋白酶,其中KLK5和KLK14能够直接降解DSG1、DSC1和CDSN;KLK7能够降解DSC1和CDSN[3]。KLK家族包括15种分泌型丝氨酸蛋白酶,其中KLK3、KLK7、KLK9和KLK15发挥糜蛋白酶样活性,其他KLK发挥胰蛋白酶样活性,只有KLK14同时具有胰蛋白酶和糜蛋白酶样作用。KLKs广泛表达于多种组织,至少11种KLKs在皮肤及其附属器中表达,主要定位于颗粒层上层和角质层[4]。KLK家族蛋白的结构高度保守,包括pre肽段(信号肽),pro肽段和active KLKs 3个部分。在角质形成细胞中,KLK以无活性的pre-pro-KLK形式生成,其信号肽在内质网被切割后,以pro-KLKs的形式通过板层小体转运到颗粒层和角质层间隙,最终通过切除pro肽段被激活而发挥酶活性。Pro肽段能够被KLKs或其他蛋白酶切割,既往研究认为KLKs通过自我激活和顺序激活其他KLKs形成蛋白水解级联反应,其中KLK5被认为是该级联反应的主要启动因素,它能够激活pro-KLK7、pro-KLK8和pro-KLK14,同时KLK14又能够激活pro-KLK5[5]。
KLKs的酶活性受到内源性抑制剂的调控。SPINK5编码的淋巴上皮 Kazal 型相关抑制剂(lympho-epithelial kazal-type-inhibitor,LEKTI)是KLK主要的内源性抑制剂,其活性片段能够不同程度地抑制KLK5、KLK6、KLK7、KLK13和KLK14的酶活性[6]。此外,还有很多其他蛋白酶抑制剂参与对KLKs活性的调控,比如SPINK9编码的LEKTI-2表达在掌跖表皮,能够抑制KLK5的酶活性;SPINK6编码的蛋白酶抑制剂也能够特异性地抑制KLK5、KLK7和KLK14的活性[7-8]。
板层小体(lamellar bodies)是一种来源于高尔基体或溶酶体的分泌性细胞器,位于棘层上层和颗粒层的角质形成细胞中,并被分泌至颗粒层和角质层间隙。其内容物主要包括脂质、蛋白酶和脂质水解酶等,研究表明板层小体能够分泌CDSN,以及KLK5、KLK7和LEKTI等[9],因此其正常形成和运输在皮肤脱屑过程中发挥重要作用。
环境因素如pH值、离子浓度和湿度也能够通过影响蛋白酶的活性影响脱屑过程。KLK-LEKTI的结合依赖于pH值,角质层的pH值从内向外梯度下降,使得KLKs在表皮最外层才能发挥功能[10]。另外,湿度也会影响KLKs的表达水平和酶活性,在干燥环境中,KLK7表达水平显著下降,导致皮肤出现鳞屑[11]。
2 脱屑异常相关遗传性疾病
脱屑和角化过程相互平衡以维持正常的角质层厚度,很多遗传性皮肤病都可以通过干扰皮肤生理脱屑过程致病。
2.1Netherton综合征 Netherton综合征(Netherton syndrome,NS)是一种SPINK5基因突变导致的常染色体隐性遗传性皮肤病,以先天性鱼鳞病样红皮病、套叠性脆发和特应性体质为特征,其中特应性体质包括特应性皮炎、哮喘、血清IgE升高和血嗜酸性粒细胞增多等[12-13]。皮损处病理表现为明显角化过度伴角化不全,颗粒层减少和棘层肥厚,以及真皮乳头层血管周围淋巴细胞浸润。
SPINK5基因突变会导致LEKTI缺乏或活性下降,造成KLK5、KLK7和KLK14等丝氨酸蛋白酶的酶活性不受抑制,从而导致角质桥粒的过度降解,表现为过度脱屑和皮肤屏障严重破坏。SPINK5基因敲除(SPINK5-/-)小鼠皮肤屏障严重受损,出生后不久死亡,同时敲除KLK5和KLK7(SPINK5-/-KLK5-/-KLK7-/-小鼠)能够挽救其临床表型和致死性[14-16];过表达KLK5(Tg-KLK5)小鼠也出现和NS患者相似的角质层剥脱、鳞屑和皮肤炎症[17]。提示KLK5、KLK7的过度激活在NS的发病中有关键作用。KLK5的过度激活能够通过蛋白酶激活受体2(PAR2)通路-核因子Kappa B(NFκB)通路或其他通路引起Th2细胞的发育和Th2细胞因子的上调[18-19]。Tg-KLK5小鼠研究中发现Th2型和Th17/Th22型炎症因子的上调[17],说明KLK的失调参与NS患者的过敏和炎症反应。
2.2SAM综合征 SAM综合征(severe skin dermatitis,multiple allergies and metabolic wasting syndrome,SAM syndrome),一种表现为严重皮炎、过敏和代谢紊乱的遗传病,呈常染色体隐性遗传或常染色体显性遗传[20]。皮肤异常主要为先天性红皮病、全身皮肤剥脱、条纹状掌跖角化病及毛发稀少,其他系统异常包括反复的食物过敏、皮肤和呼吸系统感染、嗜酸细胞性食管炎、生长受限和血清IgE水平升高等。
DSG1的纯合突变或复合杂合突变是SAM综合征的主要致病机制。DSG1杂合突变携带者仅表现为掌跖角化[21]。DSG1主要定位于棘层上部和颗粒层中的桥粒,以及角质层中的角质桥粒。DSG1的缺失导致棘层以上角质形成细胞连接异常和桥粒受损,造成皮肤屏障受损,皮损处组织学观察可见棘层松解,同时出现皮肤过度脱屑。同时,DSG1可通过抑制ERK通路促进角质形成细胞分化,其缺失可能导致角质形成细胞的过度增殖和分化抑制[22-23]。
2.3PLACK综合征 PLACK综合征(PLACK syndrome)是临床表现为泛发性皮肤剥脱、白甲、肢端点状角化、唇炎和指节垫的常染色体隐性遗传病。皮损处病理改变主要为棘层松解、表皮内裂隙和过度角化。其致病基因为CAST,编码钙蛋白酶抑制蛋白(calpastatin,CAST),一种内源性钙蛋白酶抑制剂。CAST功能丧失性突变可导致钙蛋白酶的过度激活,可能会导致桥粒结构蛋白表达异常或者过度降解,诱发角质形成细胞过度凋亡,同时引起角质形成细胞中间丝蛋白异常,细胞间黏附缺陷,从而出现水疱或皮肤剥脱。PLACK综合征为本研究组于2015年在国际上首次命名并确定及发病机制[24],全球仅有11例患者被报道[25]。
2.4皮肤剥脱综合征 皮肤剥脱综合征(peeling skin syndrome,PSS)是一组异质性的、以皮肤浅层无痛性剥脱、黏膜脆性不增加为特点的常染色体隐性遗传病。主要分为肢端皮肤剥脱综合征(acral peeling skin syndrome,APSS)和泛发型皮肤剥脱综合征(generalized peeling skin syndrome,GPSS),GPSS又分为非炎症型(PSS-A)和炎症型(PSS-B)[26]。
APSS由TGM5或CSTA突变导致,主要表现为局限于手足的皮肤剥脱,常伴红斑,皮损处组织病理检查显示过度角化和角质层与颗粒层分离。TGM5编码转谷氨酰胺-5(transglutaminase 5),催化角质包膜蛋白的交联,CSTA编码的胱抑素A(cystatin A)是角质包膜的重要前体蛋白,两者异常都会影响角质包膜的正常形成和组装[27-28]。
PSS-A由SERPINB8或FLG2突变导致,临床表现为出生后不久就出现的全身无痛性皮肤剥脱,不伴有炎症性改变。皮损处组织病理检查可见轻度角化过度、颗粒层变薄、角质层与颗粒层分离或角质层内有裂隙。SERPINB8编码的丝氨酸蛋白酶抑制剂肽酶抑制因子B支成员8(serpin8),其功能丧失性突变导致DSG1的表达增加,细胞间黏附的机械稳定性异常[29];FLG2编码的丝聚蛋白2(Filaggrin 2)是皮肤角化和屏障形成的必要成分,近年研究认为FLG2可能通过和CDSN相互作用,在角质细胞黏附中发挥功能[30]。
PSS-B在出生时或儿童早期发病,临床特点为泛发的皮肤剥脱伴红皮病、剧烈瘙痒和特应性表现。皮损处组织病理检查可见角质层下裂隙和真皮浅层轻度炎性浸润。CDSN是其致病基因,其编码的角膜锁链蛋白是角质桥粒的重要结构蛋白。CDSN基因突变后角质桥粒形成受损,角质细胞间粘连受损,造成皮肤屏障功能障碍,对外界变应原和有害物质的抵御能力下降,导致患者角质层剥脱、特应性等表现[31]。
2.5KLK11基因突变导致一种脱屑障碍性MeDOC 上述疾病均为皮肤脱屑过度伴皮肤屏障功能受损的疾病。有意思的是,当角质桥粒蛋白降解受限时,也会导致皮肤脱屑异常,表现为皮肤脱屑延迟或者脱屑障碍。2023年初,笔者在一个常染色体显性遗传家系和两个散发病例中,首次描述了一种新的皮肤红斑角化伴脱屑障碍性单基因遗传病,暂且称为常染色体显性遗传性孟德尔角化病(MeDOC,Mendelian disorders of cornification),并确定其致病基因为编码组织激肽释放酶11(KLK11)的KLK11基因[32]。
该病的临床特点为出生后不久就出现的红斑角化性斑块,并逐渐累及全身,可表现为红斑角化性斑块或者全身弥漫性红皮伴角化,可伴有黏着性鳞屑和轻度瘙痒,在干燥寒冷的环境中加重(图1a~1c)。患者不伴有其他系统异常,疾病呈现完全外显的常染色体显性遗传模式。患者皮损处病理检查显示非特征性改变,包括角化过度、角化不全和明显的棘层肥厚,以及真皮浅层轻微的血管周围单核淋巴细胞浸润。值得注意的是,患者角质层高度增厚、致密、紧实,失去篮网状结构改变(图1d)。透射电镜观察可见患者角质层外层的角质桥粒的数量明显增多,且结构完整,提示角质桥粒未被正常降解,患者角质层脱落障碍。通过全外显子组测序和Sanger测序技术确定KLK11的两个杂合突变位点c.149G>A(p.Gly50Glu)和c.148G>A(p.Gly50Arg)分别为3个家系患者的致病突变。
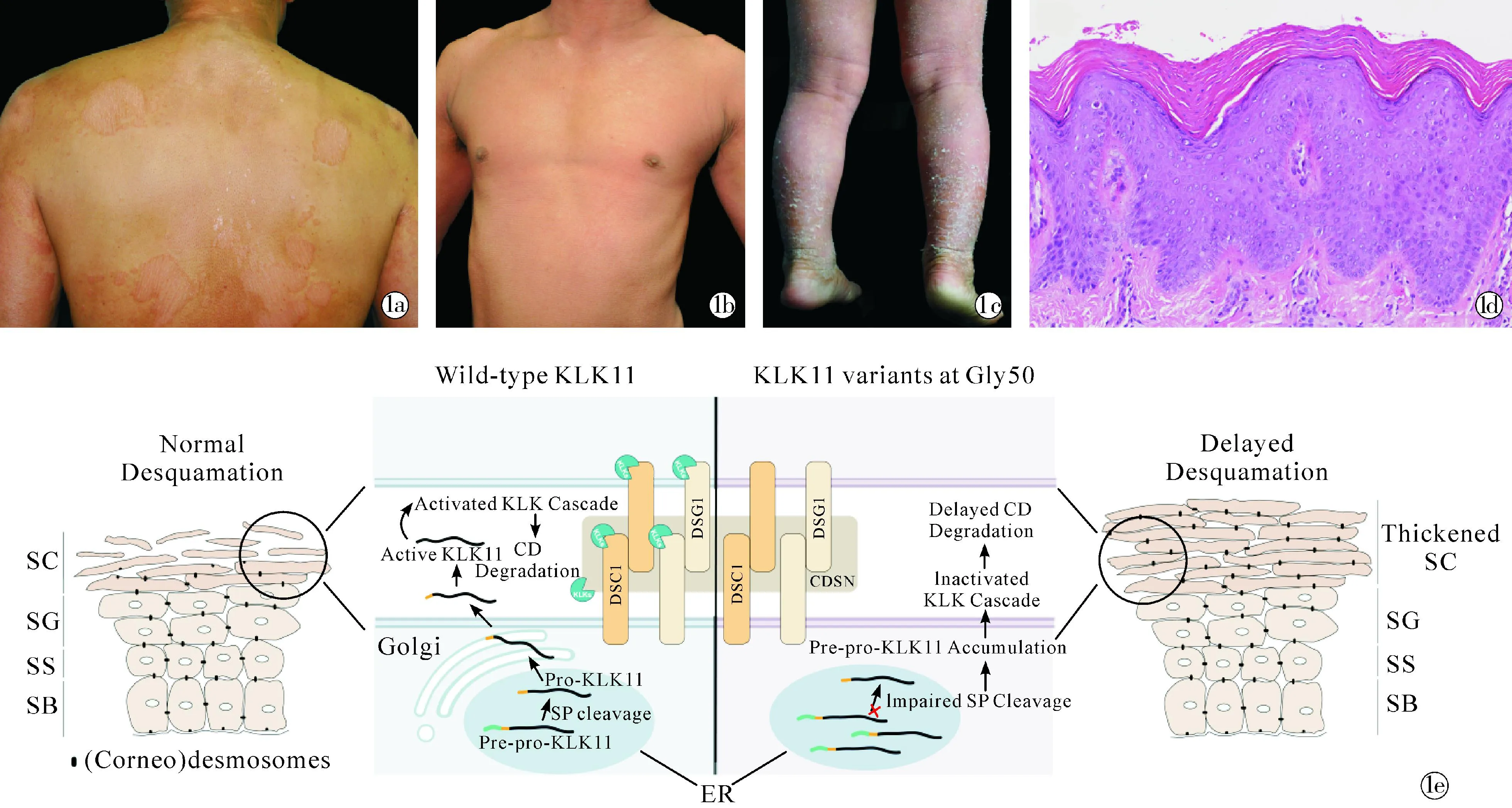
~ Erythematous hyperkeratotic plaques in individuals with Zhu-type erythrokeratodermia; HE staining of skin sections showing parakeratosis,lamellar hyperkeratosis,and acanthosis,scale bars:100 μm; The schematic illustration of Zhu-type erythro keratodermia′s pathological mechanism
KLK11属于KLK家族,发挥胰蛋白酶样活性,主要表达于表皮颗粒层上层和角质层。和其他KLK家族蛋白一样,KLK11以无活性的pre-pro-KLK11形式在角质形成细胞中合成,需经历信号肽在内质网中被切割、分泌至颗粒层和角质层间隙、以及pro肽段被切除等几个过程而发挥酶活性。既往体外研究KLK11在KLKs的蛋白水解级联反应中,能够激活pro-KLK14;KLK5、KLK6和KLK14也能够激活pro-KLK11;但其生理底物和在KLK蛋白水解级联反应中的准确功能仍未明确[3]。
患者携带的两个突变位点(p.Gly50Glu,p.Gly50Arg)影响的氨基酸Gly50位于KLK11信号肽切割位点的-1位,通过针对突变体的体外功能学实验和患者角质层的酶学分析及角质桥粒结构分析等,笔者发现患者携带的突变位点导致 KLK11 的信号肽无法被正常切割,使得 KLK11滞留在内质网,无法分泌至细胞间隙,导致角质层中 KLKs 水解级联反应的激活异常。患者表皮的胰蛋白酶和糜蛋白酶活性下降,角质桥粒成分DSG1、CDSN等的降解受阻,导致角质桥粒在外层角质中保留,角质无法正常脱落,从而阐释了KLK11突变患者出现脱屑障碍的重要发生机制(图1e)。同时,在通过CRISPR-Cas9方法构建的等效基因突变KLK11-G44E 小鼠模型中(小鼠KLK11-Gly44 位点与人类KLK11-Gly50 同源),纯合突变小鼠出生6 d开始出现明显角质层增厚伴脱屑障碍,皮肤屏障功能未受损,成功再现了患者的皮肤表现。
笔者确定了KLK11的蛋白水解位点突变可以导致一种角化异常性疾病,这是第一个KLKs家族基因突变导致的遗传性皮肤病。
据此,笔者提出该疾病的诊断标准:①出生后不久出现境界清楚的红斑角化性斑块,伴有黏着性鳞屑和轻度瘙痒,皮损可累及全身出现红皮病样改变;可伴毛发异常及鱼鳞病样皮损;②皮肤组织病理表现为表皮角化过度,可伴有角化不全、棘层肥厚、角质层紧实致密;③皮肤超微改变为角质层外层的角质桥粒的数量明显增多,结构完整;④呈常染色体显性遗传模式,存在KLK11基因杂合突变。
MeDOC不是一种确定的疾病名称,而是多种角化异常性皮肤病,包括鱼鳞病、掌跖角化、红斑角化等数十种疾病的统称。由于北京大学第一医院皮肤科朱学骏教授是首位接诊并且描述2例本病患者临床特征的医生,在本病的致病基因研究中给予了关键的方向性指导,为了避免MeDOC带来的命名混乱,因此笔者建议本病命名为“朱学骏红斑角皮病(Zhu-type erythrokeratodermia,ZEK)”,作为第一个以中国皮肤科医生名字命名的疾病[33]。
3 总结与展望
皮肤生理脱屑过程是维持皮肤屏障结构和功能稳定的关键一环,而SPINK5突变导致的Netherton综合征、DSG1突变导致的SAM综合征和CDSN突变导致的PSS-B等以过度脱屑为主要表现的单基因遗传病的发现和机制研究,为生理脱屑的发生机制,以及各种结构蛋白、蛋白酶和内源性蛋白酶抑制剂在脱屑过程中的功能提供了重要的研究模型。笔者首次报道并阐明一种KLK11基因突变导致的MeDOC,为进一步研究KLKs在皮肤角化及脱屑中的功能提供了一个突破口,将有助于皮肤屏障机制、相关疾病诊疗的研究和发展。
