Ir和IrPd催化剂的合成及结构调控对其甲醇氧化性能的影响
2024-02-23周新文沈晓宇严烙意田贵贤张荣华
周新文 沈晓宇 付 策 王 攀 严烙意 程 征 田贵贤 张荣华
(1三峡大学材料与化工学院,宜昌 443002)
(2湖北三峡实验室,宜昌 443002)
Iridium (Ir) is a member of platinum precious metals, and its catalytic performance is excellent.At the same time, it also has the shortcomings of scarce surface reserves and high cost, which lead to extremely high actual commercial costs.According to the survey,the number of reports on Ir and its related metal materials in the Web of Science database has also increased in the last decade, but the number of documents and the degree of research is much lower than other popular precious metals, also leading to the lack of comprehensive and in-depth research on Ir related systems[1].With the increasing expectations of researchers for the oxygen evolution reaction (OER) of Ir nanoparticles(NPs) used as electrocatalysts in electrolytic cells,more and more attention should be paid to research on Ir NPs[2-3].Reports on the application of Ir in various fields also show that different research groups have different strategies and methods for the synthesis of Ir NPs, and most of them try to synthesize Ir NPs by changing conditions based on existing mature methods for the synthesis of Ir NPs[4-5].This is more because there is little research on the mechanism of Ir NPs formation.At present, researchers are not clear about the path of formation of Ir in the solid phase, liquid phase, and gas phase, and because Ir NPs can often exist relatively stable with a size less than 2 nm and are not easy to agglomerate, which makes existing methods for studying the reaction kinetics and mechanism based on the optical characteristics of NPs ineffective and difficult to effectively study the formation mechanism[6].
The transformation process from Ir complex to cluster structure has been investigated.When the ligand is carbonyl, the Ir4cluster can be easily obtained[7].In the process of reducing Ir NPs by ultraviolet irradiation, many different conditions were compared and studied,such as change in precursors,different solvents, solvent volume, alkali concentration, temperature,time,and atmosphere[6].However,the reduced Ir NPs were all about 1.0-1.7 nm in nano size, and the ethanol synthesis rate was found to be much faster than that of methanol, and the NPs yield also increased under the condition of alkali ethanol.Sodium citratemodified Ir NPs (Cit-Ir NPs) were prepared by reduction of NaBH4, which had enzyme-like activity[8].Hollow iridium nanotubes (Ir NTs) with rough and porous surfaces were successfully synthesized by a selftemplate method induced by 1-hydroxyethyl ethylene-1,1-diphosphonic acid under hydrothermal conditions[9].The unique one-dimensional and multidimensional structure of Ir NTs makes them have a large surface area, high conductivity, and the best atomic utilization efficiency.Therefore, compared to commercial Ir nanocrystals (Ir-NCs),the catalytic activity and durability of Ir NTs for acidic OER are significantly enhanced, and the current density of 10 mA·m-2can be provided only by an overpotential of 245 mV.Cai′s research group[10]proposed a high-density Ir monoatomic catalyst supported by CoOxamorphous nanosheets (ANSs).The experimental results show that the single Ir atom on the CoOxANSs carrier will be anchored by many O absorbed on the surface to reach a stable state.Ir/CoOxANSs had excellent OER performance and the initial overpotential was less than 30 mV.InsituX-ray absorption spectrum (XAS)analysis shows that Ir-O-Co effectively improves the OER activity and enhances the stability of monoatomic Ir.
The triblock copolymer has been widely studied in the synthesis of noble metal catalysts due to its low toxicity, environmental friendliness, special structure,and reduction selectivity[11-16].In our previous work, it has been proven that P123 (poly (ethylene oxide)-poly(ethylene oxide)-poly (ethylene oxide) (PEO20-PPO70-PEO20)can be used as both a reducing agent and a protective agent to prepare Pd nanospheres[10], PtPd hollow nanochain[12-13]and AuPd[14-15]catalysts.We also found that IrPd catalysts with adjustable structure and MOR activity can also be obtained under the KI and P123 morphology regulators[16].At the same time, the phenomenon that Ir precursors are not easy to completely restore was also found.In this paper, the effects of water and ethanol as solvents on the morphology,structure, and MOR properties of Ir and IrPd catalysts were further studied.
1 Experimental
1.1 Chemicals and materials
Iridium (Ⅲ)chloride was purchased from Bidepharm Co., Ltd.Potassium hexachloropalladate (Ⅳ)and P123 were purchased from Sigma-Aldrich Co., Ltd.Ethanol (99.8%, GR) was purchased from Macklin Co.,Ltd.
1.2 Preparation of the Ir and IrPd nanocatalysts
In typical hydrothermal synthesis, 0.03 mmol of IrCl3and 1.0 g of P123 were dissolved in 20 mL of ultrapure water and completely dissolved by ultrasonic stirring.The obtained solution was transferred to a reactor and reacted at 180 ℃for 12 h.The resulting sample was centrifuged and washed and labeled Ir-H.Under other conditions being constant,CH3CH2OH was used instead of H2O as the solvent,and the resulting sample was labeled Ir-S.Furthermore, as a reference, 0.03 mmol of IrCl3was dissolved in 20 mL of CH3CH2OH solvent for the reaction, and the obtained sample was labeled Ir-E.
For IrPd, 0.03 mmol of IrCl3and 0.03 mmol of K2PdCl6were dissolved in 20 mL of CH3CH2OH solvent containing 1.0 g of P123 and completely dissolved by ultrasonic stirring.The above solution was reacted in a reactor at 180 ℃for 12 h,and the obtained sample was recorded as IrPd-S.IrPd-H,IrPd2-H,and Ir2Pd-H catalysts can be obtained by replacing CH3CH2OH with aqueous solution and changing the ratio of Ir and Pd under the same other conditions.To eliminate the effect of the residual protective agent P123 on the electrocatalytic performance of the products, all the catalysts obtained above must be washed three times each with water and ethanol[13-16].
1.3 Catalysts characterizations
X-ray diffraction (XRD)patterns were recorded on Rigaku (Ultima Ⅳ) with CuKαradiation (λ=0.154 06 nm).The tube voltage, tube current, scanning range,and scanning speed of XRD test were 40 kV, 40 mA,20°-90° and 4 (°)·min-1, respectively.Transmission electron microscope (TEM), high - resolution TEM(HRTEM), and high angle annular dark field scanning TEM (HAADF-STEM) were used on instruments of the JEOLJEM-F200 (acceleration voltage: 200 kV) including energy dispersive spectrometer (EDS) accessories,to investigate the morphology, structure, and composition of the products.The X-ray photoelectron spectroscopy (XPS) experiment was carried out by the AXIS Supra (Shimadzu, Japan).The UV-Vis spectrum was acquired on the UV-3600 iPlus model UV-Vis near infrared spectrophotometer(Shimadzu,Japan).
1.4 Electrochemical measurement
Cyclic voltammetry (CV) experiments were carried out on a CHI660E electrochemical workstation using a traditional three-electrode system, in which a Pt sheet (1 cm×1 cm) was used as a counter electrode(CE) and a saturated calomel electrode (SCE) was used as a reference electrode (RE).A certain amount of aspreparation IrPd catalyst was added to the clean glassy carbon (GC) electrode surface and then 10.0 μL (mass ratio of 0.5%) Nafion solution was dropped onto the electrode surface to obtain the working electrode (GC).All electrochemical experiments were performed with ultrapure water.
2 Results and discussion
TEM images, HRTEM images, and particle size distribution histograms of Ir-H, Ir-E, and Ir-S NPs are shown in Fig.1.As can be seen in Fig.1a-1c, the Ir-H NPs were random flocculent structures with slight agglomerations.The Ir-E (Fig.1e, 1f) and Ir-S (Fig.1i, 1j)NPs were highly dispersed spherical structures with average diameters of about 170 and 560 nm, respectively.The results of the enlarged TEM images showed that the three Ir catalysts were assembled by some small Ir particles.The HRTEM results showed that the crystal surface spacing of Ir-H (Fig.1c), Ir-E (Fig.1g),and Ir-S (Fig.1k) NPs was 0.224, 0.221 and 0.221 nm,respectively,which corresponds to the(111)crystal surface of Ir (PDF No.06-0598).The average particle size of Ir-H (Fig.1d), Ir-E (Fig.1h), and Ir-S (Fig.1l) NPs prepared by different synthesis methods was 2.06,1.74, and 1.93 nm, respectively.The Ir NPs of about 2 nm can exist stably[6].
The UV-Vis spectra of the IrCl3precursors in different solvents (water and ethanol) before and after the reaction are shown in Fig.S1a (Supporting information).Before the reaction, the absorption curves of the solution almost coincide, indicating that there is no significant difference between the aqueous precursor solution and the organic phase[18].After the reaction,the absorption curve of the organic phase decreased overall, but the absorption curve of the organic phase was higher than that of the organic phase, which showed that the reduction reaction of the organic phase was more thorough.The solvothermal reduction efficiency was higher and the effect was better after the hydrothermal solvent was converted to ethanol (Fig.S1b).The supernatant of Ir-H was colored and transparent, indicating that the Ir precursors were not completely reduced under the same conditions.The supernatant of Ir-S was a colorless and transparent solution (Fig.S1c), which further indicates that the reduction effect of the Ir precursor by the solvothermal method was better than that by the hydrothermal method.
Since the MOR activity of pure Ir is very low[16],we further studied the effects of hydrothermal and solvothermal methods on the synthesis and MOR performance of the IrPd catalyst.Fig.2a shows the TEM image of the IrPd-H catalyst.It can be seen from Fig.2a.that the IrPd-H catalyst is composed of some obvious nanochains, and this chain structure should be caused by the special chain structure of P123 itself[12-16].In the enlarged TEM (Fig.2b) showed that these chain structures were assembled by some hollow spheres with brighter middle and darker sides, which may be caused by a galvanic replacement reaction[19].This hollow chain structure catalyst has been shown to contribute to the improvement of its electrocatalytic performance[20-21].The crystal plane spacing obtained from HRTEM (Fig.2c) was approximately 0.232 nm,corresponding to the (111) crystal plane of the IrPd alloy catalyst.When the content of Pd increased, the IrPd2catalyst still maintained a hollow chain structure(Fig.3c and 3d), but when the content of Pd decreased,the Ir2Pd catalysts did not have a chain structure, but a disordered aggregate of some small NPs (Fig.3e and 3f).At the same time, it can be observed that relatively obvious chain structures can be observed in both IrPd2-H and IrPd-H catalysts.With the further increase in Ir content,the Ir2Pd-H catalyst was mainly of some aggregates rather than chain structures.The main reason is that for pure Ir,P123 tended to form spherical particles(Fig.1), and with increasing Ir content, the chain structure of PdIr gradually transferred to the sphere with a disordered aggregate.

Fig.2 TEM(a,b),HRTEM(c),SAED(d),HAADF-STEM(e)images,and Ir(f),Pd(g),and overlay of Ir and Pd(h)mapping images of IrPd-H catalyst

Fig.3 TEM images of catalysts IrPd-S(a,b),IrPd2-H(c,d),and Ir2Pd-H(e,f)
The results of selected area electron diffraction(SAED) (Fig.2d) showed that the obtained IrPd catalyst is a typical face-centered cubic (fcc) structure, and the diffraction rings from inside out correspond to the crystal planes (111), (200), (220), and (311), respectively.This conclusion is consistent with the results of powder XRD.The distribution of elements in IrPd-H catalysts was further investigated.The results of HAADF-STEM images (Fig.2e-2h) showed that the Pd and Ir elements in the IrPd-H catalyst were distributed throughout the product, but the distribution was not uniform.The uneven distribution of such elements may be caused by the difficulty of complete reduction of Ir precursors in this hydrothermal condition.
The particle size of the IrPd-S catalyst was larger,and the agglomeration was more obvious.Large surface particles cover many small NPs.There is a clear interface between these small particles and large particles(Fig.3a and 3b).On the other hand, the distribution of Pd and Ir elements in the IrPd-S catalyst is relatively uniform and it can be observed that the Ir elements are mainly concentrated on the surface (Fig.S2).Although the distribution of Ir and Pd elements in IrPd-S is relatively uniform, the subsequent electrocatalytic test results showed that the MOR performance of the IrPd-S catalyst was very poor.Therefore, we further analyzed the surface composition by using HADDF-STEM and element line scanning.Fig.S2 further confirms that IrPd-S was a structure of small particles that covered large particles.In Fig.S2, the element line scanning results corresponding to the scratch position in the HADDF-STEM image region are shown in Fig.S2b.It can be concluded that the overall structure of the catalyst is an IrPd alloy core with an Ir-rich layer wrapped with a higher proportion of Pd atoms[22].This is consistent with the image in the TEM image, which further confirms that the IrPd-S catalyst has an IrPd@Ir coreshell structure.
Fig.4 shows XRD patterns of IrPd-H,IrPd-S,theoretical IrPd alloys, pure Pd, and pure Ir catalysts.Both IrPd-H and IrPd-S catalysts exhibited good crystallization degree,which is a typicalfccstructure.The diffraction peaks near 39.4°, 45.9°, 66.4°, and 80.3° correspond to the crystal planes (200), (200), (220), and(311) of the IrPd alloy catalyst[16].The diffraction peaks of these diffraction angles correspond to the diffraction rings from the inside out in SAED, which further verifies itsfccstructure.Compared with the diffraction peak angles of theoretical IrPd alloy, pure Pd, and Ir catalysts, the characteristic diffraction peak angles of IrPd-H and IrPd-S catalysts had obvious negative shifts.The decrease in the diffraction angle in XRD will lead to an increase in the lattice spacing, which means that the doping of Ir atoms causes the expansion of the palladium lattice, and the catalyst forms an alloy structure[16].The reason why the diffraction angle of the IrPd alloy moves in the low angle direction is inconsistent with the reported movement between Pd and Ir[23]and in the high angle direction[24]may be that the element distribution in our prepared IrPd alloy is not too uniform.It can be seen from the dashed lines in Fig.4 that the angle corresponding to the diffraction peak of IrPd-H moves more toward the lower angle than that of IrPd-S, indicating that more Ir is incorporated into the Pd lattice in IrPd-H.It is also further proved that IrPd-S has an Ir-rich core-shell structure on its surface.

Fig.4 XRD patterns of IrPd-H and IrPd-S catalysts
Fig.5a shows the survey spectrum of the IrPd-H catalyst, confirming the coexistence of the elements Ir and Pd.Fig.5b and 5c are the spectra of the Pd3dand Ir4felements after fitting, respectively.In Fig.5b, the XPS peaks at 335.44 and 340.72 eV are Pd3d3/2peaks that correspond to zero-valent Pd[16,25], indicating that Pd is completely restored.In Fig.5c, the binding energies of 61.20 and 64.20 eV belong to Ir4f7/2and Ir4f5/2of the metal Ir[16].Peaks at 61.80 and 64.60 eV are characteristic peaks of Ir(Ⅰ)ions[26].This analysis combined with the semiquantitative characteristics of XPS shows that P123 can basically reduce Pd precursors to obtain zero-valent Pd, while only a small part of the Ir precursors are reduced to the metallic Ir, and a large part of Ir ions are still not reduced to the metallic state.

Fig.5 Survey(a),Pt4f(b),and Ir4f(c)high-resolution XPS spectra of IrPd-H
Fig.6a shows the CV curve of the IrPd series catalysts prepared by hydrothermal reaction in 1.0 mol·L-1NaOH.Among them, the CV curves of IrPd-H and IrPd2- H showed characteristics similar to those of commercial Pd-black, and the adsorption and desorption peaks corresponding to H and the redox peaks corresponding to Pd were relatively obvious.With increasing of Ir content, the characteristic peaks corresponding to Pd were weakened, and more CV characteristics of Ir were displayed[16].The CV curves of the IrPd-H series catalysts and Pd-black in 1.0 mol·L-1NaOH+1.0 mol·L-1CH3OH are shown in Fig.6b.Due to the stronger adsorption capacity of the CH3OH molecules on the catalyst surface, the peaks of adsorption and desorption of H was almost completely inhibited between -0.9 and -0.7 V.The oxidation peak of CH3OH was obvious in the forward scan at around -0.1 V and in the reverse scan at around -0.4 V.The MOR activity of the IrPd-H and IrPd2-H catalysts was higher than that of commercial Pd-black catalysts, while the MOR activity of the Ir2Pd-H catalyst was lower than that of Pd-black due to the high inert Ir element.The statistical results of the forward scanning oxidation peak potential (EP), forward scanning oxidation peak current density (jP), negative scanning oxidation peak potential (EN), and negative scanning oxidation peak current density(jN)of different IrPd catalysts are shown in Table 1.Furthermore,the ratio ofjPtojNcan be used to briefly evaluate the ability of the catalyst to resist CO poisoning[21].The higher the value ofjP/jN, the stronger the ability of the catalyst to resist CO poisoning.As shown in Table 1,while the IrPd-H catalyst showed the highest MOR activity, it also showed stronger resistance to CO toxicity.Table S1 compares the MOR performance of IrPd catalysts in recent years.The MOR performance of the IrPd catalyst we obtained still has great room for optimization and improvement.
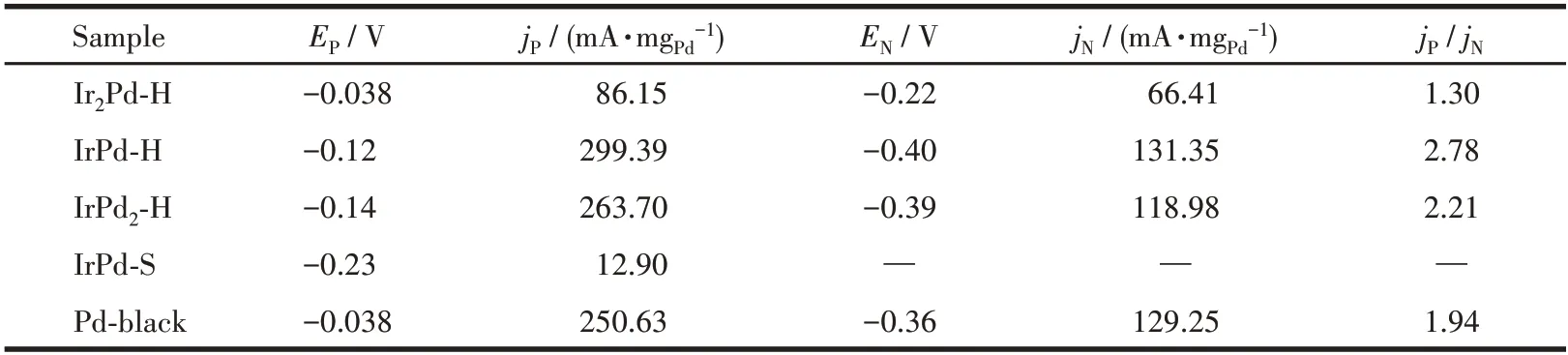
Table 1 Main electrocatalytic data obtained from Fig.6

Fig.6 CV curves of IrPd-H,IrPd2-H,Ir2Pd-H,and Pd-black catalysts in(a)1.0 mol·L-1 NaOH and(b)1.0 mol·L-1 NaOH+1.0 mol·L-1 CH3OH;(c)MOR performance comparison of IrPd-H,IrPd-S,and Pd-black catalysts
Fig.6c shows the CV curves of the MOR performance comparison of IrPd-H,IrPd-S,and Pd-black catalysts.The IrPd-H catalyst had good MOR catalytic activity,and its maximum oxidation peak current density reached 299.39 mA·mgPd-1, which was 1.2 times that of the commercial Pd-black catalyst.At the same time,the potential corresponding to the highest oxidation peak of the palladium black catalyst also shifted negatively from -0.038 to -0.12 V.However, the MOR activity of the IrPd-S catalyst was much lower than that of IrPd-H and Pd-black catalysts.The main reason is that the surface of IrPd-S is mainly inert Ir elements,which further verifies that the IrPd-S catalyst is a coreshell structure with an Ir-rich surface.The core-shell structure of the IrPd-S catalyst can be changed by introducing a morphology regulator, and the catalytic activity of MOR can be greatly improved[16].
3 Conclusions
In this paper, the effects of hydrothermal and solvothermal methods on the synthesis of pure Ir and IrPd catalysts under the action of P123 were discussed based on the regulation of structure and surface composition of precious metal Ir-based catalysts.For pure Ir catalyst, the solvothermal methods of ethanol solution with a certain reduction ability as solvent are helpful to improve the reduction efficiency, and the Ir particle size of 1.93 nm was obtained with a uniform distribution.Under the same conditions, the traditional hydrothermal reaction can obtain Ir self-assembled nanospheres with a particle size of about 2 nm, but the reduction was not complete.For binary IrPd catalytic systems, core-shell IrPd@Ir catalysts with very low MOR activity were obtained by the solvothermal method.The hollow chain IrPd catalyst was prepared by the hydrothermal method, which showed MOR activity higher than that of commercial Pd black.
The results show that for Ir-based catalysts,different synthesis methods can effectively adjust the degree of reaction,the morphology,structure,and surface composition of the product,and it is expected to be extended to other Ir-based catalysts and catalysis fields.
Acknowledgments:The financial support from the National Natural Science Foundation of China (Grants No.21503120, 21403126), and the 111 Project (Grant No.D20015)are highly acknowledged.
Supporting information is available at http://www.wjhxxb.cn
