多相催化加氢反应中H2异裂解离的研究进展
2024-02-02姚正阳王晓月郭晓宏赵勇杰师文荣李聪明
姚正阳,王晓月,郭晓宏,赵勇杰,师文荣,李聪明
(太原理工大学 省部共建煤基能源清洁高效利用国家重点实验室,山西 太原 030024)
催化剂在化学反应中具有选择性加速作用,能够使反应快速达到平衡状态或选择性促进某一产物的生成,其中,多相催化在规模化放大以及产物分离等方面具有明显优势。在众多高附加值化学产品的生产过程中,均涉及加氢步骤,如通过加氢反应将含不饱和化学键(C≡≡C、C==O、C==N 以及N≡≡N 键等)的物质转化为高附加值化学品(烯烃、芳烃、胺、汽油、醇以及氨气等)[1]。与H2均裂过程相同,H2异裂解离产生的氢化物(M—Hδ-)广泛参与均相催化反应[2]、H2/CH3OH储存[3]、非均相催化反应[4]、光催化[5]和电催化[6]等反应。更重要的是,研究发现H2异裂解离产生的M—Hδ-对C==O、C==N和N==O键等极性键具有选择性加氢的特点,为高选择性合成目标产物提供了新途径[7]。因此,了解并认识H2异裂解离形成的M—Hδ-对加氢反应机理的影响规律是十分必要的。然而,多相催化剂复杂的表面结构导致H2异裂解离过程和M—Hδ-的化学性质复杂多变,为理解H2异裂解离过程与催化剂结构的内在关系及其对加氢反应机理的调控规律带来了巨大挑战。
本文综述近期在多相催化剂表面和体相中H2异裂解离过程及其M—Hδ-的化学性质对反应机理的影响,以加深对多相催化剂体系中H2异裂解离以及M—Hδ-的认识。首先,概述H2异裂解离机理和M—Hδ-反应性。其次,介绍研究M—Hδ-的主要表征技术及其各自优缺点。随后,讨论不同催化体系中M—Hδ-对加氢反应活性和选择性的影响以及调控M—Hδ-化学性质的策略。最后,总结当前研究进展的局限性,对未来研究方向进行展望。
1 H2异裂解离机理及氢化物的性质
1.1 H2异裂解离机理
H2分子的活化,即H—H键的断裂,往往需要借助催化剂以扰动成键电子对,最终形成具有化学活性的中间体。然而,具有不同性质的催化剂会使H2发生不同的解离,主要包括H2均裂解离以及H2异裂解离,见图1。当H2分子靠近催化剂并吸附在其表面时,金属中心的d轨道与H2的分子轨道发生相互作用(如金属中心的d轨道转移电子到H2分子的反键轨道),实现H—H键的断裂。当金属中心邻近配位的原子仍是金属时(M—M 键),通常会形成两个相同性质的金属—氢键(M—H 键),即H2遵循均裂机理。但是,在多相催化剂中,金属中心的配位环境往往具有复杂且多样化的特点。通常,金属中心(Lewis酸)会与O、S、N或P等非金属原子(Lewis碱)进行配位形成Lewis 酸碱对。当H2分子吸附在Lewis 酸碱对上时,在亲核原子或强电场的作用下极化并解离,其中与金属中心成键的H带有负电荷形成M—Hδ-,而与非金属原子成键的H带有正电荷形成质子氢(B—Hδ+),即H2遵循异裂解离机理[8],以O为例,H2异裂解离过程见式(1)。
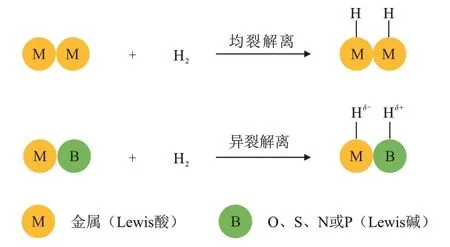
图1 H2的均裂解离和异裂解离[8]Fig. 1 Homolysis and heterolysis of H2[8]
根据Lewis 酸碱对的类型(具有不同金属—非金属成键性质),H2异裂解离形成的M—Hδ-和质子氢(O—Hδ+)具有丰富、可调的化学性质,可以更好的满足不同反应基元步骤的需求,在决定加氢反应活性和选择性中具有重要作用。因此,相比于H2均裂解离,H2异裂解离在多相加氢反应过程中占有重要地位。
1.2 氢化物的类型和化学性质
H2在催化剂表面的异裂解离需要在亲核原子或强电场的作用下使H2极化并分裂。金属自身的原子半径、电离能和电负性等性质的差异会导致对H2的极化和成键能力不同,其中电负性作为衡量形成化学键时吸引电子能力强弱的指标,是最为显著的影响因素之一[9]。根据电负性的差异(表1),氢化物的类型大致可分为3 种:(1)碱金属与碱土金属,由于自身电负性较低,当与H结合时容易向外转移电子,氢原子获得一个电子成为H-生成的离子型氢化物;(2)对于大多数d 区和f 区元素能形成金属型氢化物,而VⅠB族仅有Cr能形成金属型氢化物;(3)对于后过渡态金属以及非金属元素,电负性与氢接近,在其表面更倾向于以共价键形成M—H物种。以电负性1.35作为分界,所有直接形成氢化物(离子型与金属型氢化物)的金属电负性都小于1.35。

表1 电负性与氢化物类型的关系及对应的元素[9]Table 1 Relationships between electronegativity with hydride types and corresponding elements[9]
通常在反应中,主要活性氢物种是由异裂解离产生的离子型氢化物。受氢原子自身电荷的影响,离子型氢化物有利于C==O、C==N 和N==O 等极性不饱和键加氢,这也为加氢反应选择性的调控提供了可操作平台。XⅠANG等[10]通过简单的沉淀-还原法制备的以CoO 为壳层的Co@CoO 催化剂,经H2异裂解离得到大量稳定的Hδ-物种,在5-羟甲基糠醛(HMF)制备2,5-二甲基呋喃(DMF)的反应中,Hδ-物种优先加氢至极性不饱和的C==O 键,从而保护了HMF 中的C==C 键,在130 ℃下DMF 产率达到89.2%。LⅠU等[11]制备了单原子分散Pd1/TiO2催化剂用于光催化加氢反应,研究发现该催化剂在C==C键和C==O 键的加氢反应中都表现出良好的活性,催化苯乙烯和苯甲醛加氢反应的转化频率(TOF)分别是商业Pd/C催化剂的9 倍和55 倍。理论计算结果表明,Pd1/TiO2通过异裂解离路径活化H2,生成了Pd—Hδ-,因此催化剂在反应中表现出对不饱和键超高的加氢反应活性。以上结果表明,合理设计催化剂,调控H2的解离方式,可为在加氢反应中高选择性制备目标产物提供新途径。
2 研究H2异裂解离的主要表征技术
多相催化剂活性中心复杂的化学性质和配位环境使M—Hδ-具有多样性,为提高加氢反应活性和选择性提供了机会,但同时也为认识“催化剂结构-氢化物性质-反应机理”三者之间的内在关系带来了挑战。重要的是,研究三者之间的内在关系用以指导设计高效催化剂是十分必要的。当前,已有较多光谱表征手段用于研究多相催化体系中H2异裂解离机理、M—Hδ-的形成及其相关作用。以下简要介绍常用研究M—Hδ-的表征技术以及M—Hδ-在反应过程中的作用。
2.1 1H核磁共振光谱
1H核磁共振光谱(1H NMR)可表征物质含氢物种的种类及相对含量,当H2在催化剂表面发生异裂解离会随之产生M—Hδ-和B—Hδ+。因此,利用1H NMR可表征催化剂表面是否存在M—Hδ-和B—Hδ+,并可将表征结果作为判定H2是否发生异裂解离的重要依据。
WU等[12]制备了MgO与Ru/MgO催化剂并分别进行1H NMR 检测,结果见图2。MgO(111)表面出现表面羟基(化学位移0.71)和极化的MgO—H(化学位移5.43)两个信号峰。对于Ru掺杂的MgO(111),化学位移-1.7 的主信号峰归因于极性MgO(111)上的Ru—H,化学位移-4.8 的信号峰归属于Ru—Hδ-。当Ru 掺杂MgO(111)后,形成Ru—O—Mg 界面,其中界面处的O2-作为Lewis 碱为H2的反键轨道提供了电子,而H2的成键电子与作为Lewis 酸的Ru2+相互作用,最终加速H2异裂解离。这类似于均相受阻Lewis 酸碱对(FLPs)可逆活化H2的作用机制[13-14]。
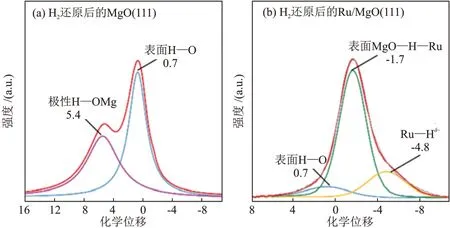
图2 H2 还原后MgO(111) (a)和Ru/MgO(111) (b)的1H NMR光谱[12]Fig. 2 1H NMR spectra of MgO(111) (a) and Ru/MgO(111) (b)after H2 reduction[12]
CHEN 等[15]通过对H2活化前后1H 魔角旋转固体核磁共振谱(1H MAS NMR)进行差值处理,定量研究H2活化过程中Ga2O3表面氢物种的形成。结果表明,在Ga2O3表面同时发生H2的均裂解离和异裂解离,分别产生两个相邻的Ga—OH 基团以及一对Ga—Hδ-和Ga—OH 基团。更重要的是1H NMR 结果定量分析发现,Ga—OH和Ga—Hδ-形成的峰面积比值接近3∶1,表明H2在Ga2O3表面发生均裂解离和异裂解离概率相同。因此,1H NMR 表征技术是可定量化研究H2解离方式的重要手段之一。
2.2 红外光谱
H2在催化剂表面进行解离时,往往会改变催化剂表面原子的电子结构,最终导致催化剂表面形成的氢物种振动峰的强度以及频率的变化(即红外信号的变化)。因此,根据M—Hδ-和O—Hδ+振动峰的存在及变化可以判断催化剂表面是否存在H2的异裂解离以及性质,同时可利用同位素D2解离进一步证实。COPERET 等[6]通过红外光谱(ⅠR)观察到了在不同金属氧化物表面H2异裂解离形成的M—Hδ-。由于ZnO、ZrO2和CeO2在加氢反应中应用较为广泛,因此以该3种氧化物为例,对红外光谱在加氢反应过程中的应用和研究进行了介绍。
GARCÍA-MELCHOR等[16]与NEGREⅠROUS等[17]通过对H2与CeO2(111)相互作用的机制进行研究,发现CeO2(111)表面的Ce4+和O2-分别作为Lewis 酸碱对促使H2发生异裂解离。WERNER 等[18]为了明确CeO2能作为高效加氢催化剂的本质原因,利用红外反射吸收光谱,在不同温度以及压力条件下,研究了具有不同氧空位密度的CeO2(111)薄膜与H2的相互作用机制,同时对CeO2(111)表面和体相的OD(OH)基团的比例以及振动频率进行理论计算,结果见图3。实验和理论计算结果共同表明,CeO2(111)上的氧空位可以稳定H2异裂解离形成M—Hδ-和羟基物种。

图3 D2 气氛下CeO2(111)和CeO2-x(111)薄膜的IR 光谱((a)~(d))及CeO2(111)表面和体相OD (OH)基团的比例振动频率(e)[18]Fig. 3 IR spectra of CeO2(111) and CeO2-x(111) thin films exposed to D2 ((a)~(d)) and scaled vibrational frequencies of surface and bulk OD(OH) groups on CeO2(111) (e)[18]
另外,LⅠ等[19]通过使用红外光谱,首次研究了CeO2(111)表面和体相氢化物M—Hδ-形成过程的区别,结果证明,氧空位浓度对氢物种形成与稳定发挥了重要作用。总的趋势为随着氧空位浓度的增加,O—Hδ+趋于不稳定,而M—Hδ-趋于稳定。另外,表面O—Hδ+比体相O—Hδ+更稳定,而体相M—Hδ-比表面M—Hδ-更稳定。有趣的是,GARCÍA-MELCHOR等[20]发现H2在CeO2表面的异裂解离产生的氢物种不仅可以参与加氢反应,还可以限制炔烃分子的吸附和扩散来进一步阻止低聚物的形成,从而提高炔烃加氢的活性和选择性。
ZnO 因其具有优良的H2解离能力被广泛的用于加氢反应[21],关于ZnO 表面氢物种的红外光谱也有较为深入的研究。KOKES等[22]发现,H2在ZnO表面异裂解离产生的两种红外信号峰位于1705 cm-1和3490 cm-1,分别归属于Zn—H 和O—H 物种的伸缩振动。将D2替换H2后,新的红外信号峰出现在1225 cm-1和2585 cm-1,分别归属于Zn—D 和O—D物种的伸缩振动。除此之外,BOCCUZZⅠ等[23]与HUSSAⅠN 等[24]研究发现,在817 cm-1、850 cm-1处也会分别出现Zn—H与O—H的伸缩振动峰,同时也在1475 cm-1处发现了以Zn—H—Zn形式存在的M—Hδ-。最近DONG 等[25]对添加Cr 的ZnO 表面氢物种的形成和演变进行了研究,H2在其表面的解离路径见图4。除Zn—D(1230 cm-1)和O—D(2580 cm-1)外,还发现(Cr—)Zn—D(1300 cm-1)和(Cr—)O—D(2700 cm-1)的特征峰存在,结合密度泛函理论(DFT)计算结果证明非极性ZnO(101¯0)和极性ZnCr2O4(111)表面活性位点性质的改变会影响催化剂表面M—Hδ-和羟基的性质,最终导致高温加氢反应性能的变化。

图4 H2在ZnO与ZnCrOx表面的解离路径[25]Fig. 4 Dissociation path of H2 on ZnO and ZnCrOx surface[25]
虽然ZrO2是一种难还原的氧化物,然而在低温条件下仍会形成两种氢化物。ONⅠSHⅠ等[26]利用傅里叶变换红外光谱研究了CO 加氢反应过程中,ZrO2催化剂表面H2的解离行为部分结果见表2。由表2 可知,H2在ZrO2表面可异裂解离形成两种氢化物,即Zr—H和Zr—H—Zr,并且同位素D2替换实验进一步证明此结果。KONDO 等[27]在研究乙烯在ZrO2表面加氢的反应机理时发现Zr—H红外信号峰(1562 cm-1)和O—H 红外信号峰(3668 cm-1),表明H2在ZrO2表面发生异裂解离。不难发现,红外光谱不仅可表征催化剂表面H2异裂解离形成的氢物种类型,更重要的是,可以更好地研究已形成的氢物种对基元反应步骤的影响。
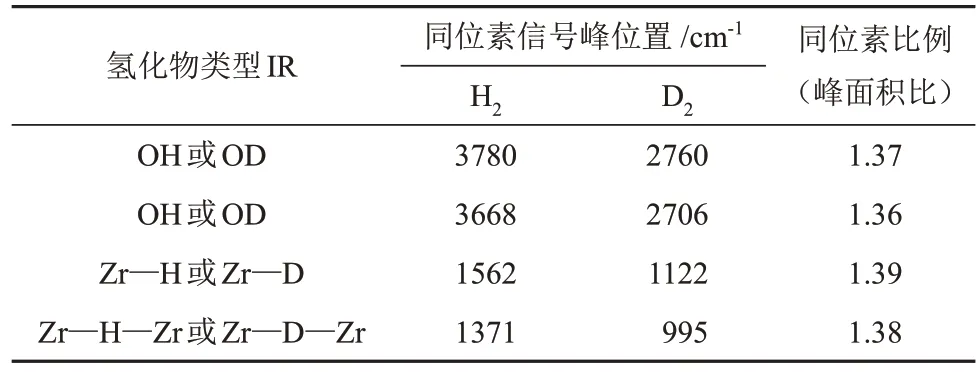
表2 ZrO2表面氢化物类型、同位素IR 信号峰位置和同位素比例[26]Table 2 ZrO2 surface hydrogen species and IR peak location of isotope and isotope ratio[26]
2.3 非弹性中子散射光谱
非弹性中子散射(ⅠNS)光谱作为可以同时研究表面和体相M—Hδ-的表征技术,已被广泛应用[28]。相比于红外光谱,ⅠNS光谱在低频范围(低于1000 cm-1)内具有更强的检测能力。此外,不同于其他元素,氢的非相干中子横截面较大,因此ⅠNS 光谱对氢物种具有较高的灵敏度。更重要的是,ⅠNS 光谱可实现量化催化剂表面和体相中的氢含量。另外,ⅠNS检测结果结合DFT 理论计算,可以更好地研究氢物种的化学环境。上述优点使ⅠNS 光谱对催化剂表面氢物种的检测具有独特的适用性。例如SⅠLVERWOOD 等[29]报道了通过H2还原Au/CeO2催化剂后,ⅠNS光谱和ⅠR光谱均检测出其表面形成了Au—Hδ-,证明H2在界面Au-O 位点处发生异裂解离[30]。WU等[31]通过ⅠNS光谱证实H2在棒状CeO2发生异裂解离行为[32]。尽管ⅠNS光谱已经被广泛用于研究催化剂表面氢物种,但较低的检测能力以及较苛刻的检测条件限制了其进一步应用。例如JUÁREZ 等[33]在研究H2、CO2和O2与Au/CeO2的相互作用时,由于催化剂样品中Au 负载量低,ⅠNS 光谱无法明显检测出表面M—Hδ-,但在400~600 cm-1处发现了羟基的吸收峰,最后结合红外光谱确定了H2在Au/CeO2表面的异裂解离行为,产生的H+吸附在载体上,H-吸附在Au 上(图5)。因此,多表征手段结合研究H2的异裂解离行为以及M—Hδ-的化学性质是未来发展方向。

图5 Au/CeO2的INS光谱((a)~(c))和Au/CeO2上H2的异裂解离(d)和CO2加氢(e)机理[33]Fig. 5 INS spectra of Au/CeO2 ((a)~(c)) and mechanism of H2 heterolysis (d) and CO2 hydrogenation (e) on Au/CeO2[33]
2.4 其他表征方法
从上文讨论中易知,H2在催化剂表面形成M—Hδ-的过程涉及电子转移,因此可以通过X射线光电子能谱(XPS)和电子自旋共振光谱(ESR)检测氧化物中金属中心和非金属元素的电子态密度变换,借此反映催化剂表面H2演变行为。LⅠ等[34]利用XPS和ESR 检测CeO2-x粉末和CeO2-x(111)薄膜吸附H2并发生还原行为后,Ce 物种价态的变化,结果见图6。还原后的CeO2-x粉末和CeO2-x(111)薄膜表面的Ce3+含量均减少。CeO2-x粉末体相中Ce3+含量同样减少,并且还原后的CeO2-x(111)薄膜其Ce3+变成Ce4+。以上结果表明,H2均裂解离会导致CeO2-x的氧化,同时形成但此方法仅能作为佐证,因此不再进行仔细论述。

图6 CeO2-x在H2处理前后(a)和逐步退火前后(b)的Ce3+含量(XPS数据所得峰面积之比)[34]Fig. 6 Ce3+ content (ratio of peak area obtained from XPS data) of CeO2-x before and after H2 treatment (a) and stepwise annealing (b)[34]
综上所述,在加氢反应中,催化剂表面或体相的M—Hδ-通常参与反应基元步骤,并发挥重要作用。但探究M—Hδ-的化学性质和结构仍然具有挑战性。目前对催化剂中M—Hδ-的表征方法主要有1H NMR、ⅠR 和ⅠNS 等。虽然以上表征技术都较为成熟,但仍有各自的局限性。例如,1H NMR无法观察到所有的氢化物种类;ⅠR和拉曼光谱具有较高的表面敏感性但较难提供M—Hδ-的定量信息;而ⅠNS的操作条件苛刻,往往需要在低温下进行测量,限制了其原位研究的能力。因此,需要通过联合多种表征技术,提供更为严谨、全面的研究结果。
3 催化H2异裂解离的催化剂类型
在实际多相催化加氢反应中,H2的异裂解离行为及其M—Hδ-的化学性质与催化剂化学性质和电子结构息息相关。然而,多相催化剂通常具有表面结构复杂的特点,导致局部配位环境和活性位点性质多样化,从而诱导不同的H2异裂解离行为,最终影响加氢反应性能。因此,总结分析催化剂微观结构与H2异裂解离行为的内在关系是十分必要的。针对负载型金属催化剂、金属氧化物催化剂以及阴离子杂化金属催化剂等体系,尝试阐述不同催化剂类型对H2异裂解离的影响以及形成的M—Hδ-对加氢反应活性和选择性的影响规律,旨在为设计催化剂结构,调控M—Hδ-化学性质,优化加氢反应性能提供理论指导。
3.1 负载型金属催化剂
当金属纳米颗粒负载于载体表面时,金属-载体界面的形成诱导金属原子的电子结构和化学性质发生改变,往往使催化剂表现出不寻常的催化性能(图7)。由图7可知,当界面金属原子的配位结构满足Lewis 酸碱对的要求时,可促使H2异裂解离在金属-载体界面处发生。负载型金属催化剂可大致分为负载型贵金属催化剂(Au、Pt、Pb、Ru和Rh等)和负载型非贵金属催化剂(Cu、Co、Fe和Ni等)两大类。
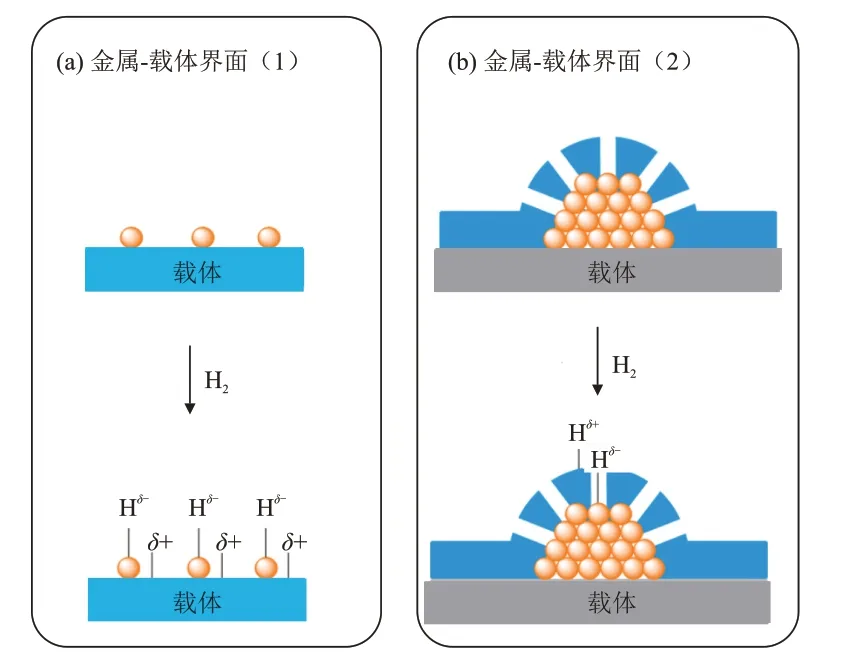
图7 两种不同金属-载体界面上的H2异裂解离[8]Fig. 7 H2 heterolysis on two different metal-support interfaces[8]
研究人员对H2在负载型贵金属催化剂上的异裂解离进行了研究,发现碱金属助剂改性能够改变催化剂的电子结构,并调控H2在催化剂表面的异裂解离能力[35]。QⅠN 等[36]对比分析了Na 改性前后的Ru/Al2O3催化剂用于丙酮加氢制备异丙醇反应。在150 °C、3 MPa的反应条件下,Na改性Ru/Al2O3催化剂的TOF达到2.2 mol/(mol·s)(1 s内1 mol催化剂转化2.2 mol丙酮),并在32 h内保持稳定。相反地,未改性的Ru/Al2O3活性衰减迅速,最终TOF仅达到0.4 mol/(mol·s)。结合DFT 计算以及原位红外光谱发现,Na 的供电子效应稳定Ru 上异裂解离的氢化物(Ru—Hδ-)。最近KⅠM 等[37]制备了Ru/MgO 用于芳烃的加氢反应,并引入碱金属K 作助剂,显著提高了Ru/MgO 的加氢活性。研究发现,碱金属K 向Ru-MgO 界面附近的Ru 转移电荷,加速H2异裂解离。电荷转移的程度取决于碱金属的离子半径和电负性,而催化剂的加氢反应活性与H2异裂解离产生的M—Hδ-含量呈正比。因此,可依据此规律,对催化剂进行合理有效设计,提高催化剂加氢活性和选择性。
除Pd、Ru 等贵金属纳米颗粒常被用作加氢催化剂外,单原子Au 在加氢反应中同样表现出加氢活性。SUN 等[38]结合DFT 理论计算对比研究了负载型Au 催化剂的尺寸效应。结果表明,相比于Au纳米颗粒,TiO2负载的单原子Au形成Au—O—Ti界面结构显著降低H2异裂解离能垒,同时H2异裂解离能垒与界面氧原子的电子密度密切相关。LⅠU等[39]通过DFT计算研究了Au/ZrO2催化剂催化丁二烯加氢反应(图8)。结果表明,形成的Au—O 界面位点作为Lewis酸碱对,促使H2异裂解离,产生Au—Hδ-和O—Hδ+物种,其中Hδ+和Hδ-依次转移与丁二烯进行加氢反应,并抑制丁烷的生成进而提高丁烯选择性。

图8 Au—H—H—Au构型中轨道相互作用的模型[39]Fig. 8 Models of orbital interactions in an Au—H—H—Au configuration[39]
由于负载型非贵金属催化剂价格低廉且活性较高,而被广泛应用于各种反应。然而,非贵金属自身H2解离能力较弱,且在实际反应条件下(如高温等),形成的M—Hδ-稳定性较差,因此很少有明确的光谱证据报道氢物种的解离及其对反应的调控作用,大多以DFT模型证明非贵金属催化剂遵循H2异裂解离机理。如LⅠ等[40]建立Al2O3负载的Ni团簇结构模型进行DFT 计算,结果表明H2在Ni 团簇表面H2异裂解离的激活势垒为0.31 eV,而在界面处的激活势垒仅为0.16 eV,证明金属-氧化物界面位点对H2的异裂解离发挥了重要作用。SARMA等[41]基于DFT 计算研究了Ti 掺杂的SnO2催化剂催化CO2选择性加氢制备甲酸的反应机理。结果表明,H2在Ti—O 键上异裂解离的能垒最低,H2异裂解离形成了带负电荷的氢化物(Ti—Hδ-),有助于CO2加氢生成甲酸。RⅠLEY等[42]基于DFT的研究,提出空间分离的O 和Ce 位点组成的受阻Lewis 酸碱对促进H2在CeO2(111)的表面上异裂解离,并基于计算结果设计了CeO2负载的单原子Ni催化剂用于乙炔部分加氢反应,其乙炔转化率超过60%。实验和理论计算结果证明,通过设计催化剂界面位点,可调控H2异裂解离能力和性质,实现优化加氢反应的活性和选择性。
以上研究结果表明,对于负载型金属催化剂,金属与载体形成的界面位点通常具有Lewis酸碱对的性质,可以加速H2的异裂解离并稳定形成的M—Hδ-。通过引入碱金属助剂、改变载体类型和性质、降低金属尺寸以及形成合金等手段,改变界面位点性质,调控其对H2异裂解离的催化作用,实现加氢反应的活性和选择性的提高。
3.2 金属氧化物催化剂
不同于负载型金属催化剂,金属氧化物催化剂表面的位点满足金属中心(Lewis 酸)与O(Lewis碱)配位形成Lewis酸碱对结构,使H2在金属氧化物表面通常以异裂解离的方式形成活性氢物种。以3 种金属氧化物(MgO、Ga2O3和Ⅰn2O3)为例,概述金属氧化物表面结构性质对H2异裂解离的作用规律。
通过红外光谱可以检测MgO催化H2异裂解离形成的O—H物种信号峰(3454 cm-1和1325 cm-1)和M—Hδ-物种信号峰(3712 cm-1和1125 cm-1)[43]。CHEN 等[44]报道了一种Ni-MgO/MgH2催化剂用于CO2还原反应发现H2异裂解离产生的H+和H-物种具有不同的反应性。CO2可通过羧酸盐路径在Ni-MgO表面与H+反应还原成CO,生成的CO 再与H-加氢生成CH4。相比于CO 经H+加氢的较高能垒(1.00~2.00 eV),CO 经H-加氢具有更低能垒(1.03 eV),可依次生成中间体HCO、H2CO、H3CO和CH4[45]。
Ga2O3催化的H2异裂解离产生的Ga—Hδ-在2003 cm-1和1980 cm-1处显示出红外信号峰,且高温条件下红外的振动吸收峰更加明显[46],DFT计算结果表明,高温可以促进氧空位的形成以稳定Ga—Hδ-[47],同时证明Ga—Hδ-能够提高CO2加氢反应活性。当Ga2O3催化逆水煤气(RWGS)反应时,红外光谱结果显示,Ga2O3表面的碳酸盐基团可以与Ga—Hδ-发生反应转化为甲酸盐物种,并进一步生成气态的CO[48]。Ga2O3用于光催化RWGS 反应时,观察到表面碳酸盐基团与Ga—Hδ-加氢生成双齿甲酸盐物种,并进一步产生CO[49]。
在Ⅰn2O3催化CO2加氢合成甲醇反应中,甲醇选择性接近100%,且研究表明H2异裂解离路径对甲醇选择性起决定性作用[50]。当H2在Ⅰn2O3表面发生异裂解离,可分别形成O—H 与Ⅰn—Hδ-。研究人员通过DFT 计算分别研究了CO2的两种可能的转化途径:(1)CO2与吸附在Ⅰn位点的H发生反应形成甲酸盐;(2)O位点吸附的H质子化CO2,形成羧酸盐。结果表明,在Ⅰn2O3表面,CO2加氢生成甲酸盐物种比质子化生成碳酸氢盐物种更有利,从而促进甲酸盐路径,提高甲醇选择性[51]。
综上所述,M—Hδ-在金属氧化物催化剂中起着重要的催化作用,而不同金属氧化物表面形成的M—Hδ-的反应性差异显著,这意味着即使在相同的加氢反应中,加氢反应路径也会有很大的不同,从而导致最终产物的差异。为了更好地理解不同金属氧化物表面M—Hδ-的反应性,需要进行进一步研究。了解金属氧化物催化剂上不同金属氧化物表面形成的M—Hδ-的性质和反应性,对于理解加氢反应机理以及优化催化剂设计具有重要意义,LⅠ等[52]提供了相关研究进行了总结,可供参考。
3.3 阴离子杂化金属催化剂
相比于金属氧化物催化剂,金属与硫、磷和氮等阴离子杂化形成的相应金属化合物(硫化物、磷化物和氮化物等)有着独特的电子结构、晶格结构、表面性能、体缺陷和金属—氧键强度等性质。更重要的是,金属与O、S、N 和P 位点满足Lewis 酸碱对标准,使得阴离子杂化金属催化剂具有H2异裂解离的能力并表现出较优异的催化加氢性能。
非贵金属中MoS2因其优异的催化剂性能而被广泛关注。HU等[53]合成了富含硫空位的二维MoS2纳米片,并用于CO2加氢合成甲醇反应,其表现出优异的低温高活性、高甲醇选择性。根据H2在MoS2-x缺陷上的异裂解离过程(图9)可知,MoS2催化剂上H2的异裂解离需要配位不饱和金属位点和相邻的硫化物离子协同作用,即Lewis酸碱对[8,54]。目前为止,通过ⅠNS证实了巯基(S—Hδ+)的形成,但并没有明确的实验证据表明Mo—Hδ-的形成[55]。

图9 H2在MoS2-x缺陷上的异裂解离[8]Fig. 9 H2 heterolysis on defective of MoS2-x[8]
在硫化贵金属催化剂中,RuS2是研究热点[55]。CAⅠ等[56]对RuS2上H+和H-在异芳烃催化加氢中的作用机理进行了研究。结果表明,H2在RuS2表面进行异裂解离,产生的M—Hδ-和质子(S—Hδ+)作为活性氢物种参与不同加氢步骤,协同完成整个催化过程。另外,关于RuS2的其余研究可见BREYSSE等[55]的总结。ALBANⅠ等[57]通过Na2S 水溶液对负载在石墨氮化碳上的Pd 纳米粒子进行处理获得Pd4S 化合物。H2在Pd4S 催化剂上通过异裂解离途径进行炔烃部分氢化反应,表现出100%的烯烃选择性。DFT计算表明,硫原子通过接受氢原子形成巯基参与加氢反应,巯基随后将氢转移到反应物中,完成催化循环。
ZHOU等[58]通过原位红外光谱并结合DFT计算发现,含P 的NbOPO4催化剂由于其表面富含氧空位,H2可以通过异裂解离和均裂解离生成稳定的M—Hδ-,不仅可以使苯基环己烷转化为单环化合物,而且可以通过加氢裂解Csp2—Csp3键使聚苯乙烯有高选择性并转化为芳烃。
WU 等[59]研究发现,MoN 表面Mo 与N 组成了H2异裂解离所需的Lewis 酸碱对结构,并通过红外光谱表征证明了H2异裂解离的反应途径(图10)。WYVRATT 等[60]将MoN 用于巴豆醛加氢制备1-正丁醇反应并通过ⅠNS 光谱证明了MoN 表面存在H2异裂解离。除此之外,利用DFT 计算研究了在巴豆醛加氢过程中M—Hδ-对产物选择性的影响。结果表明,异裂解离产生的Mo—Hδ-可选择性加氢巴豆醛中的极性C==N 键,而保留C==C 键,而高选择性合成1-正丁醇。

图10 Mo2N/Al2O3催化剂上H2的两种异裂解离方式[59]Fig. 10 Two ways for H2 heterolysis on Mo2N/Al2O3 catalysts[59]
整体而言,虽然多相催化剂表面结构复杂,但形成的具有类似Lewis酸碱对结构的活性位点,更有利于H2异裂解离,从而形成相应的M—Hδ-和O—Hδ+。通过设计催化剂结构,构建具有Lewis 酸碱对的活性位点以及通过碱金属助剂选择、载体选择、降低金属尺寸和调变金属中心的配位原子等手段优化Lewis 酸碱对活性位点的电子化学性质,进而实现调控H2异裂解离的能力及形成的M—Hδ-的化学性质和稳定性。更重要的是,H2异裂解离形成的M—Hδ-和O—Hδ+往往表现出高选择性加氢的特点[61]。一方面,在不饱和化合物中,由于极性键是M—Hδ-和O—Hδ+的良好受体,极性基团如C==O、C==N和N==O键的选择性氢化作用超过C==C 键,这为操纵加氢反应动力学和控制催化反应选择性生成目标产物提供良好的机会。另一方面,已有研究证明,异裂解离的氢物种在非极性键加氢方面也非常活跃[62]。因此,仍需更多的研究工作来阐明催化活性位点的化学性质(Lewis 酸碱对结构)、H2异裂解离能力及其M—Hδ-的化学性质与催化加氢反应性能之间的内在关系。
4 结语与展望
虽然加氢反应在制备高附加值化学品和燃料中占有重要地位且已被广泛研究,但是针对H2异裂解离行为与催化剂活性位点结构的内在关系以及其对反应机理的影响规律仍需进一步研究。本文综述了H2异裂解离机理及性质、用于表征H2异裂解离的先进表征技术以及H2异裂解离行为在不同类型催化剂上的异同及其对加氢反应性能的影响。整体而言,当催化剂金属中心(Lewis 酸)与配位原子O、S、N和P(Lewis碱)形成Lewis酸碱对时,促使H2发生异裂解离,从而产生M—Hδ-。由于M—Hδ-本身的电荷性质,能够在同时存在极性和非极性不饱和键的情况下选择性地对极性键加氢,从而实现目标产物的高选择性。通过对催化剂结构的合理设计和电子性质的调控,可以改变H2在催化剂表面的解离能力和M—Hδ-的稳定性,实现高性能的催化加氢反应。
为进一步研究并阐明H2异裂解离在多相催化加氢反应中发挥的作用及其对反应性能的影响规律,未来可从以下4 个方面开展研究:(1)设计开发更加灵敏且可定量化的表征技术;(2)多表征技术联合使用,提供更为准确全面的表征信息;(3)采用单晶的模型催化剂,降低催化剂表面结构的复杂性;(4)实验表征与理论计算相结合。随着研究的不断积累和深入,可从结构简单的单晶模型逐渐转变成较为复杂的纳米颗粒,为进一步指导高效催化剂的设计提供指导。
