金属氯化物固态电解质及其全固态电池研究现状与展望
2024-01-26程晓斌罗锦达姚宏斌
李 枫,程晓斌,罗锦达,姚宏斌,
(1中国科学技术大学合肥微尺度物质科学国家研究中心;2中国科学技术大学化学与材料科学学院,安徽 合肥 230026)
锂离子电池因高比能、高工作电压及长寿命等突出优势[1-2],已广泛应用于便携式电子设备及电气交通等领域[3]。然而,锂离子电池有限的能量密度(400 Wh/kg)[4-5]及有机液态电解质导致的潜在安全隐患等问题愈加突出[6-9],无法满足可再生新能源大规模应用的需求,这一现状迫切要求锂离子电池技术进一步突破能量密度限制,并同时提升电池体系的安全性。研究表明,实现高能量密度与本征安全的关键在于电池体系及其组成材料的物理化学性质。现阶段,电池全固态化技术从根本上避免有机液态电解质的引入和使用,在材料组成上有效保证了其本征安全性;同时固态无机化合物的热力学稳定性使得正负极材料的选择可拓展至高比容量或高工作电位的材料体系,进而大幅度提升电池的能量密度[4,10],正成为下一代锂电池的重要发展方向[11]。
然而,目前全固态锂电池的大规模应用仍然面临着循环性能差、电极-电解质界面不兼容及固-固接触不良等亟待解决的问题[7,12-17]。作为全固态锂电池的关键组成部分,锂离子固态电解质的综合性能决定了其循环稳定性、能量密度及服役寿命。因此,开发全固态锂电池的核心在于研制高离子电导率、界面稳定且易于形变的固态电解质材料[10,17-18]。目前所报道的金属氧化物、金属硫化物无机固态电解质材料各有其优势和短板,均无法同时满足全固态锂电池的应用需求。金属氧化物型固态电解质基于非移动金属阳离子及氧离子构建石榴石型[19]、NASICON型[20]、钙钛矿型[21-22]三维骨架,可移动锂离子占据其中的间隙或空位。其中,石榴石型固态电解质因其锂金属负极兼容性及成本效益得以在过去几十年内迅速发展。石榴石型的化学通式为Li3+xA3B2O12(A、B 代表高价金属阳离子,如La3+、Zr4+、Ta5+、Nb5+等)[19]。通过高温固相烧结可以实现四方对称结构和立方结构石榴石的制备,其离子传导骨架均由[LaO8]十二面体(24c)和[ZrO6]八面体(16a)构建而成,锂离子随机部分占据两类间隙位点,即48g或者偏离中心的96h位点,离子电导率一般可以达到10-6~10-4S/cm。然而由于负二价的氧阴离子半径较小(126 pm),与金属阳离子间静电作用较强,一方面会影响锂离子在本体中的传导,另一方面将会导致其晶格不易变形,无法形成接触良好的电解质-电极界面,严重阻碍界面处的离子传导,限制了基于氧化物基固态电解质的全固态电池的有效集成[23]。金属硫化物型固态电解质包含thio-LISICON型[24]、硫银锗矿型(Li6PS5Cl)[25]、锂锗磷硫型[1,26],及玻璃-陶瓷锂磷硫型[27-28],锂离子通过占据四面体位点可实现超快离子传导。硫化物固态电解质阴离子框架具有独特的堆积方式,包括体心立方(bodycentered cubic,bcc)密堆积、面心立方(face-centered cubic close packing,fcc)密堆积及六方(hexagonal close packing,hcp)密堆积[29]。其中体心立方阴离子框架由于具有能量等效的四面体位点连通网络,比其他密堆积框架具有更低的离子迁移能垒(0.15 eV)和更高的室温离子电导率(1~25 mS/cm),这在硫化物超离子导体如Li10GeP2S12和Li7P3S11中得到了验证。尽管如此,硫离子电负性较低(pauling scale,2.58),耐氧化稳定性差,因此硫化物型固态电解质材料及其全固态锂电池面临严重的界面化学/电化学稳定性问题[30],基于此产生的空间电荷层,元素扩散及高离子阻抗的电极-电解质界面,加速了全固态电池的性能衰退。可以看出,金属氧化物和金属硫化物固态电解质目前仍然存在界面稳定及离子传导之间的非理想权衡,限制了全固态电池的实用化进程。因此,通过元素化学调控,构建界面稳定且离子可导的新型固态电解质对于实现高性能全固态锂电池至关重要。
金属氯化物基固态电解质材料因具备独特的阴离子化学特性[31],兼顾了金属氧化物和金属硫化物的性能优势,实现了良好的界面稳定性与离子电导率的理想平衡(图1)[32-35]。首先,单价氯阴离子与锂离子的静电相互作用比二价的硫或氧阴离子弱,有利于实现锂离子的快速传导(10-4~10-2S/cm);同时,氯阴离子半径较大,基于氯化物的传导框架表现出较高的变形性,可实现固-固组分之间优异的界面接触;此外,氯阴离子的电负性接近氧阴离子,具备理想的氧化电位(>4 Vvs.Li+/Li),与高工作电压的正极材料表现出优异的界面兼容性,可实现高电压窗口稳定运行。研究表明,部分金属氯化物可以通过液相反应实现大规模批量生产[36-37],因此金属氯化物被视为一类综合性能优异且具有发展潜力的固体电解质材料体系。
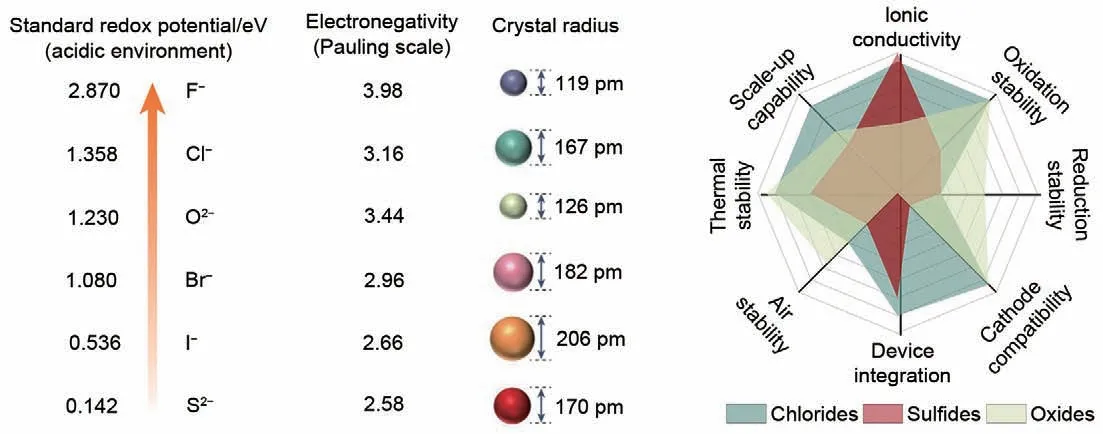
图1 金属氯化物、金属硫化物及金属氧化物阴离子化学及相应的固态电解质性能对比[31]Fig.1 Chemistry of metal chlorides, metal sulfides and metal oxide anions and corresponding properties of solid electrolytes[31]
本文结合了近五年来国内外研究机构关于金属氯化物固态电解质材料的研究成果及本课题组的一些工作进展,对其一般合成方法、晶体结构、离子传导机制、性能优化策略、电极-电解质界面演化及稳定机制,以及基于此的全固态锂电池电化学性能进行了系统的总结与评述,并对其实用化的可行性进行了分析与探讨,指出金属氯化物固态电解质材料未来进一步探索与发展的可能方向。
1 金属氯化物固态电解质发展概述
金属氯化物固态电解质材料的化学通式为Li-M-Cl,M代表非锂金属阳离子。金属氯化物的离子传导特性在20世纪90年代已经被Kanno R、Lutz H D 等课题组发现[38-43],但与当时先进的金属氧化物及硫化物固态电解质相比[44],其较低的室温离子电导率(10-7~10-5S/cm)并未引起研究者的广泛关注。2018年,日本松下电器产业株式会社Asano T等研究人员[32]提出Li3YCl6(LYC)作为新型固态电解质,其冷压粉末的室温离子电导率为0.54 mS/cm,表明金属氯化物亦可实现快离子传导。除此之外,需要特别说明的是,LYC与无保护涂层的高压钴酸锂正极材料(LiCoO2,LCO)表现出良好的化学及电化学相容性,所组装的Li-In|LYC|LCO 全固态锂电池展现了优异的循环稳定性。由此,金属氯化物固态电解质的离子电导率和全固态电池性能的突破再次引发了国内外研究者的关注。
如图2所示,2018—2023年期间,加拿大西安大略大学孙学良,滑铁卢大学Nazar L F,中国科学技术大学Ma等[32,37,45-46]研究人员及本课题组相继开发了代表性的晶态Li3MCl6(M=Y,Sc,In,Er)型、Li2MCl6(M=Zr, Hf) 型[47-49]、 Li2Sc2/3Cl4型[50-51]、SmCl3·0.5LiCl型[52]及独特的UCl3框架型固态电解质材料[53],均表现出较高的室温离子电导率(0.51~3.02 mS/cm)。实验结果表明,通过控制合成工艺,亦可制备玻璃-陶瓷型金属氯化物,有利于其室温离子电导率的进一步提升[54-55]。此外,金属氯化物晶格框架的离子容忍性和结构可设计性强,通过引入不同种类的阴离子可制备独特的金属氟氯化合物及金属氧氯化合物。2021 年韩国三星高等技术研究院Jung Sung-Kyun及2022年燕山大学张隆等研究人员分别开发了黏土状2LiCl-GaF3[56]和共晶2LiCl-0.5AlF3-0.5GaF3[57],其离子电导率分别高达3.7 mS/cm及16 mS/cm;然而,黏土状及共晶卤化物由于结晶性弱,其结构形成过程与离子传导机理尚不明确,有待进一步研究与探讨。值得一提的是,2023 年日本松下电器产业株式会社Tanaka Y 等研究人员[58]提出一类新型氧氯化物——LiTaOCl4,继承了卤化物的氧化稳定性和形变性,表现出可媲美液态电解质的室温离子电导率(12.4 mS/cm),基于此的全固态电池展现了优异的充放电特性,证明了金属氯化物这一新兴材料体系在全固态锂电池产业化进程中的巨大潜力。
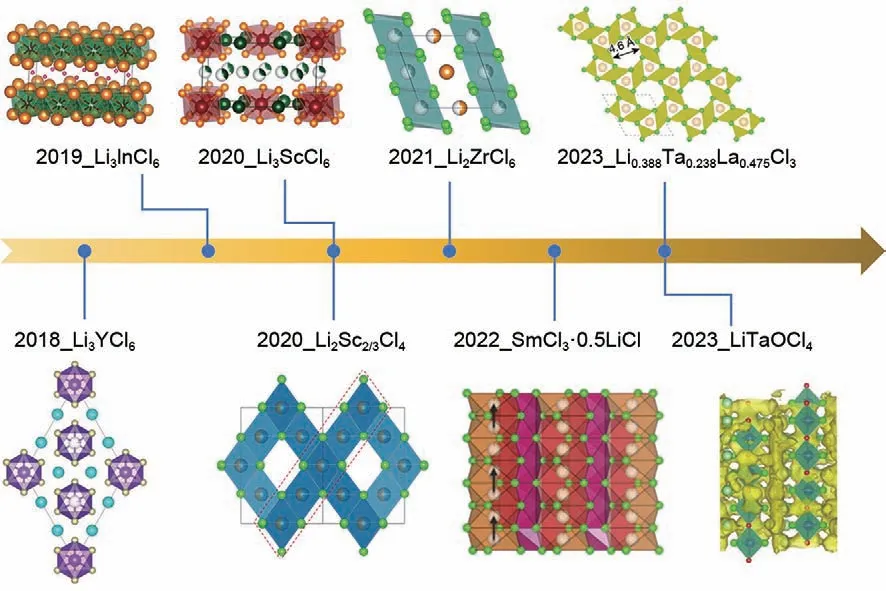
图2 近五年来代表性的金属氯化物固态电解质研究进展[32,37,45,48,51-53,58]Fig.2 Representative development of MCSEs in recent five years[32,37,45,48,51-53,58]
2 金属氯化物固态电解质的合成方法
现阶段,金属氯化物固态电解质的合成主要依赖于固相反应和液相反应。固相反应合成法的优点在于工艺流程简洁,只需将混合均匀的氯化物原料通过高能球磨或高温共熔烧结,即可得到目标产物。虽然这种方法适用范围广,但在大批量合成时,产品的均匀性和一致性还有待进一步优化。因此,固相反应合成法更适合于实验室规模新材料的研发和初步性能的验证。另外,液相反应合成法是在液相环境中进行均匀反应,然后通过高温处理去除剩余的溶剂,从而得到金属氯化物固态电解质。这种方法具有大规模生产的潜力,但在选择溶剂和后处理方式时,通常会受到合成原料物理化学性质的限制。因此,目前这种方法还不具备普遍适用性。
2.1 固相法
固相反应合成(图3)是高能球磨或高温共熔烧结,或是两者结合使用。高能球磨法[59]是通过球磨珠、罐的内壁之间的相互碰撞产生的机械能供给化学反应的活化能,制备质地均匀的粉末,产物多为低结晶度或玻璃-陶瓷亚稳态材料。这类材料通常具有较多的本征缺陷或较高的结构无序度,可有效降低离子迁移激活能,因此表现出较高的室温离子电导率。现阶段,基于高能球磨法已经成功实现了一系列高离子电导率的Li3MCl6(M=Y,In,Er,Yb)材料的制备与应用[32,47-48,60-61]。此外,德国吉森大学Schlem R等研究人员[46]通过结构精修分析发现,高能球磨法制备的Li3YCl6及Li3ErCl6较高温烧结法得到的离子电导率高出一个数量级的原因在于高能球磨导致其晶体结构中产生了丰富的阳离子缺陷,诱导锂亚晶格重排,通过扩展锂扩散瓶颈通道提升其离子扩散速度。不同的工艺所产生的性能差异表明合成方法学在实现高性能固态电解质合成中具有重要作用。
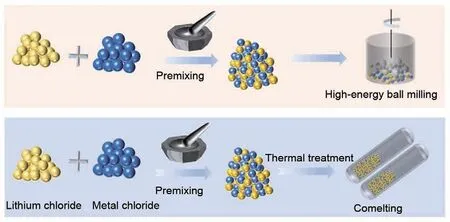
图3 金属氯化物固态电解质固相反应合成法(球磨、烧结)示意图[36]Fig.3 Schematic of solid phase synthesis of metal chloride solid electrolytes (ball milling, sintering) [36]
高温共熔烧结法,通常是将混合均匀的原料进行高温熔融处理,随后对其缓慢降温,析出热力学稳定相,其产物一般具备较高的结晶度和较大的粒径。此外,通过控制降温速率可以制备高质量的高结晶度样品,适用于晶体结构表征[45,50]。
2.2 液相法
液相反应合成法(图4)通常采用合适的溶剂,将原料完全溶解。通过自然干燥或热处理方式,获取水合物前驱体。最终,通过高温加热彻底去除结晶水及吸附水,即可得到目标产物。然而,由于氯化物原料对湿度极为敏感,这一方法在某些情况下缺乏通用性。现阶段,加拿大西安大略大学的孙学良等研究人员[37]成功展现了水相介导合成的Li3InCl6离子电导率为2.04 mS/cm,略高于固相反应合成法所得材料的离子电导率(1.4 mS/cm)。尽管如此,由于稀土基Li3MCl6(M=Y,Er,Sc)在水相溶液中不可逆地降解为LiCl、MCl3及MOCl,无法通过后续的加热处理获取原始的金属氯化物。为了进一步推动液相反应合成法的广泛应用,孙学良等研究人员[31]采用了铵盐辅助液相合成的创新工艺,通过形成稳定的中间体[NH4]3[MCl6],结合热处理方式,成功实现了Li3MCl6(M=Y,Er,Sc)的液相法合成,突破了水相介导法的限制。尽管盐辅助液相法在大规模合成方面具有潜在的优势,但所得的产物离子电导率低于传统球磨法所得的离子电导率,其衰退机理目前仍需要进一步深入研究和澄清。
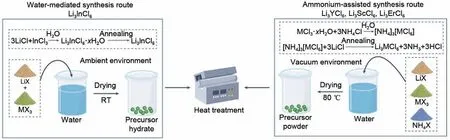
图4 金属氯化物固态电解质液相反应合成法示意图[31]Fig.4 Schematic of liquid phase synthesis of metal chloride solid electrolytes [31]
综上,金属氯化物固态电解质的制备方法对其离子传输性能、粒径大小及均匀性的影响至关重要。因此,选择实用高效且具备成本效益的金属氯化物固态电解质材料合成技术对于推进全固态锂电池应用进程具有重要的研究意义。
3 金属氯化物固态电解质的晶体框架
实验与理论结果表明,传统的Li-M-Cl的晶格框架可以认为是以LiCl作为基本结构,通过引入高价金属阳离子进行“掺杂”替换,伴随着空位缺陷产生,从而构建快速的导锂框架[图5(a)]。根据阴阳离子密堆积原则[62],即当阴阳离子半径比值满足稳定配位数规则时,即可形成特定的密堆积亚晶格。诸如基于阴阳离子六配位形式的六方密堆积(hexagonal close packing,hcp)亚晶格及立方密堆积(cubic close packing,ccp)亚晶格,明显不同于硫化物固态电解质中的体心立方密堆积(body-centered cubic,bcc)亚晶格[28]。此外,本课题组开创性地提出一种基于镧系稀土氯化物UCl3型晶格框架的固态电解质材料体系,镧系稀土离子与氯离子具有独特的“三帽三棱柱”九配位亚晶格,完全不同于传统的四配位或者六配位的金属氧化物/硫化物固态电解质的结构单元及密堆积的金属氯化物结构单元。以下分别对其进行详细介绍。
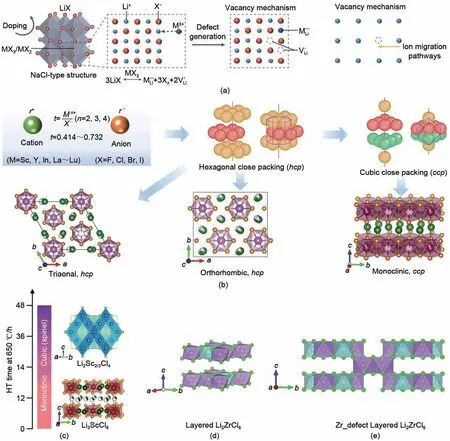
图5 (a) Li-M-Cl晶体框架及空位形成机理[31];(b) 基于阴阳离子半径之比的原则所得的六方及立方密堆积晶格[64];(c) Li-Sc-Cl立方密堆积单斜结构及尖晶石结构[45,50];(d) 基于神经网络势函数原子模拟得到的理想Li2ZrCl6层状结构[49]及(e) 含锆缺陷的Li2ZrCl6层状结构[49]Fig.5 (a) Formation mechanism of Li-M-Cl crystal framework and vacancy[31; (b) hexagonal and cubic close packed lattice based on the principle of the ratio of the radius of anions and ions[64]; (c) cubic close-packed monoclinic structure and spinel structure of Li-Sc-Cl[45,50]; (d) the ideal Li2ZrCl6 layered structure[49] and (e)Li2ZrCl6 layered structure with zirconium defects obtained by atomic simulation[49]
3.1 Li-M-Cl型晶体框架
金属氯化物Li3MCl6体系中阳离子Li+、Sc3+、Y3+、Er3+、Yb3+及In3+的半径[63]分别是90 pm、88.5 pm、104 pm、103 pm、100.8 pm 及94 pm,与氯阴离子的半径(167 pm)之比均在0.414~0.732。根据Pauling 离子晶体稳定规则[62]可知,可形成稳定的[LiCl6]及[MCl6]六配位八面体,通过边共享或面共享等方式相连,进而构建三维导锂框架[图5(b)]。常见的结构包括基于六方密堆积的三方结构(P-3m1),其中非锂阳离子M3+的离子半径分布范围为102~106.4 pm,可能的阳离子包括Tb3+、Dy3+、Ho3+、Tm3+、Er3+及Y3+;基于六方密堆积的正交结构(Pnma),其中非锂阳离子M3+的离子半径分布范围为100~102 pm,可能的阳离子包括Lu3+及Yb3+;基于立方密堆积的单斜结构(C2/m),其中非锂阳离子M3+的离子半径小于100 pm,可能的阳离子包括Sc3+及In3+。需要特别指出的是,尽管通过Pauling离子晶体稳定规则可预测一系列Li3MCl6固态电解质材料,但是,目前经过实验成功制备的仅有Li3ScCl6、Li3YCl6、Li3ErCl6、Li3YbCl6及Li3InCl6。我们分析这可能是由于其他类型的Li3MCl6并不具有热力学稳定相,其中涉及的热力学稳定机理仍然需要更多理论模拟与相关的实验表征去揭示。值得注意的是,同组分配比的Li-Sc-Cl在同一温度下经过不同时间的退火处理,分别形成立方密堆积单斜结构[45]和尖晶石结构[50][图5(c)]。研究人员认为是由于其形成能相近,通过控制反应条件,即可实现晶体结构转变。
近期,Li2MCl6(M=Zr,Hf)体系因其可观的离子电导率、湿度稳定性及成本效益优势,引起国内外研究者的广泛关注[47-49]。研究表明,Li2MCl6体系的晶体结构和离子电导率与其合成途径密切相关。通过对比X 射线衍射谱发现,高温烧结所得的Li2ZrCl6展示了类似于Li3InCl6的ccp单斜结构,但表现出极低的离子电导率(<10-5S/cm);而高能球磨法所得的结构类似于Li3YCl6的hcp六方结构,并展示出更好的离子传输性能(10-3S/cm)。然而,需要说明的是,通过X 射线衍射谱的对比分析而提出的结构,是否可以作为Li2ZrCl6的真实结构尚有争议,需要通过更多的结构表征与数据分析进一步验证。此外,本课题组通过对Li-Zr-Cl 体系进行神经网络势函数原子模拟,高通量筛选得到(LiCl)1-x(ZrCl4)x的热力学凸图以及全局最稳定相Li2ZrCl6。全局优化结果揭示了Li2ZrCl6热力学稳定相中的锂离子与锆离子均与氯离子采用六配位模式,构成LiCl5-6和ZrCl2-6八面体,这种配位模式理论上符合鲍林离子晶体排列规则,基于边共享的八面体搭建富含本征空位缺陷的新型层状结构[图5(d)、(e)]。
3.2 UCl3型晶体框架
在氯化物晶体框架中,如果非锂阳离子半径进一步增大,则阴阳离子配位方式将从传统的六配位过渡到八配位或九配位模式。早在1994年,Lissner F等研究人员[65]就在Na3xM2-xCl6(M=La~Sm,P63/m)这类材料中发现了这一现象。具体而言,MCl3形成P63/m晶体框架,且沿c轴方向有一维(1D)空位贯穿通道。其中,部分钠离子占据了M位点,其余钠离子则分布于通道空位,丰富的八面体空位提供了较短的离子跳跃距离,理论上可以实现钠的快速扩散。然而,所报道的钠离子电导率仍远低于实际应用的要求(>10-4S/cm),限制了该类框架型材料在快离子导体开发中的进一步开发与利用。
2023 年,本课题组从离子导体的阴离子框架构建原理出发,提出具有独特结构的镧系稀土氯化物基固态电解质材料的设计与制备[53]。不同于传统的六方密堆积(hcp)及立方密堆积(ccp)阴离子框架,UCl3型阴离子非密堆积晶体框架的结构单元为镧氯九配位形式的“三帽三棱柱”,突破了传统的四配位、六配位结构基元的框架设计。在镧系稀土氯化物的晶体框架内,具有很多天然存在的大尺寸一维孔道(4.6 Å),可供锂离子占据并提供快速迁移位点,有利于降低锂离子迁移势垒。同时,一维孔道内有很多未被占据的扭曲八面体位点,减小了相邻位点间的扩散距离,位点与位点之间的距离较为接近(2.2 Å),局部相似的配位环境提供了平滑的能量曲面以及连续的渗流网络,非常适合离子进行快速扩散(图6)。此外,加拿大西安大略大学孙学良等研究人员[52]亦发现一类基于SmCl3的类沸石拓扑氯化物骨架,其中一维通道被[SmCl9]6-三棱柱包围,在两个八面体之间提供2.08 Å 的短跳跃距离用于锂离子的跳跃。此外,SmCl3框架可以与传统金属氯化物固态电解质复合,以获得迁移载流子,证明了界面结合行为和离子扩散在UCl3框架材料中的普遍性。
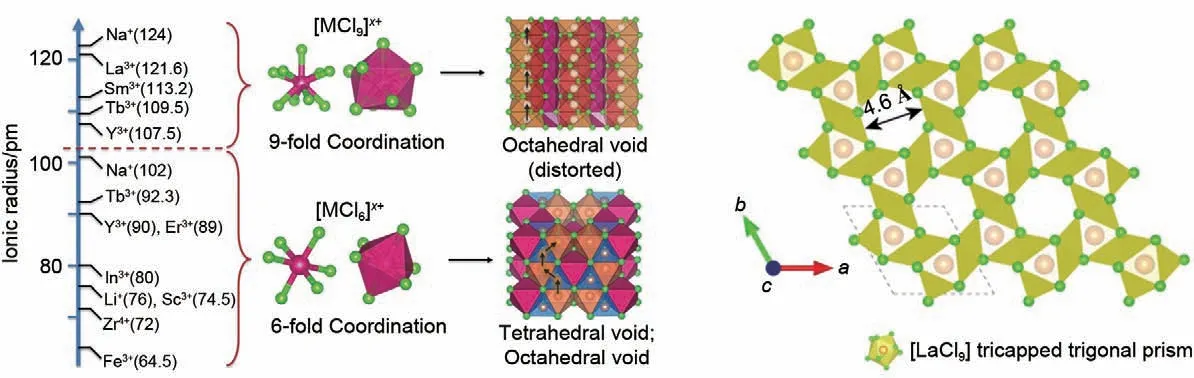
图6 UCl3型晶体结构框架[52-53]Fig.6 Framework of UCl3-type metal chlorides[52-53]
4 金属氯化物固态电解质的离子传导机制
不同于传统的金属硫化物、金属氧化物固态电解质中锂离子的迁移机制,金属氯化物固态电解质不需要依赖体心立方(bcc)的阴离子亚晶格与多离子协同跃迁机制,其锂离子迁移是通过具有低势垒的本征空位点在多面体位点之间的跳跃传输而实现。基于上文所述可知,金属氯化物晶体结构由六方密堆积、立方密堆积或非密堆积的UCl3型亚晶格组成,其中,Li+通过与Cl-进行六配位,形成[LiCl6]八面体,并占据其中心位点。理论模拟结果表明,在不同的氯离子亚晶格中,锂离子稳定占据态、迁移过渡态及其周围的亚晶格环境不尽相同,因此离子传输路径及其对应的迁移势垒明显不同,主要传导机制阐述如下[32-33,45]。
金属氯化物通过在阴离子堆叠层之间的离子键实现锂离子的快速迁移(图7)。在六方密堆积氯离子亚晶格(hcp_Li3YCl6)中,锂离子通过在c轴方向上跳跃至两个面共享的八面体(Octahedral,Oct)位点,形成了快速的一维扩散通道(被称为Oct-Oct路径)。这些Oct-Oct 路径通过四面体(Tetrahedron,Tet)间隙在ab平面上相互连接(被称为Oct-Tet-Oct路径),构建了一个各向异性的三维扩散网络。然而,晶体结构中常见的反位缺陷、杂质和晶界可能会引发“阻塞效应”,从而影响c轴方向一维扩散通道中锂离子的快速传导。在立方密堆积氯离子亚晶格(ccp_Li3ScCl6)中,锂离子通过在两个边共享的八面体位点之间的四面体间隙跳跃,形成了各向同性的三维扩散通道(被称为Oct-Tet-Oct路径)。同时,ccp和hcp氯离子亚晶格中的锂离子迁移活化能明显低于ccp和hcp氧或硫离子亚晶格,这表明ccp和hcp阴离子亚晶格更适合用于开发金属氯化物固态电解质材料。
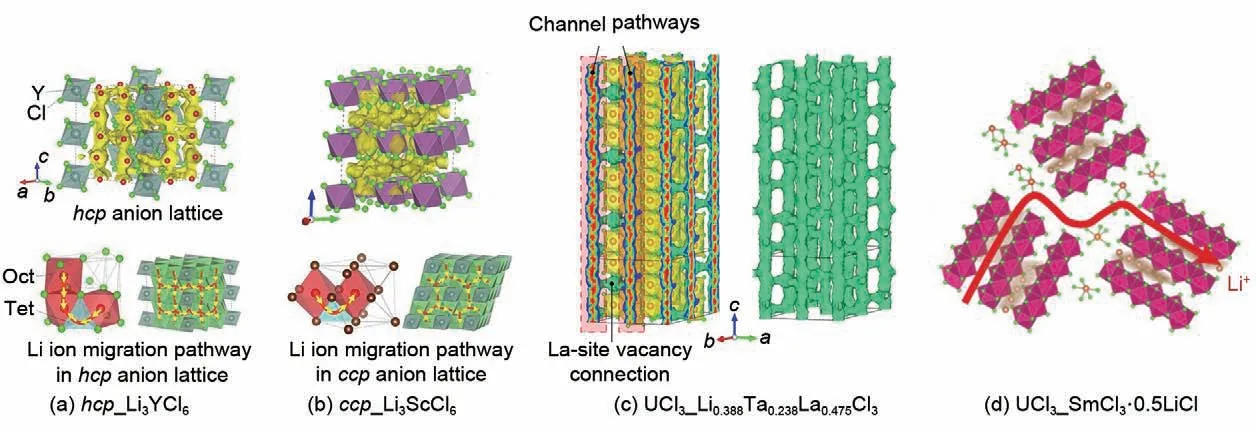
图7 金属氯化物固态电解质离子迁移机制[45,52-53,64]Fig.7 Mechanism of ion migration in metal chloride solid electrolyte[45,52-53,64]
在UCl3镧系稀土氯化物晶格中,氯阴离子以非紧密堆积的方式排列,形成了丰富的一维大尺寸孔道。此外,通过镧空位的形成,可以实现连续的三维传导,使锂离子能够在整个晶格中自由移动。本课题组首次发现,在LaCl3型晶格中,通过引入高价金属离子(如W6+、Ta5+、Nb5+、Zr4+、Hf4+)可以制造大量的镧空位。这些镧空位触发了大尺寸高速离子通道和相邻通道间的超强交换作用,从而构建出三维贯通的离子渗流通道。具体而言,锂离子在该通道中通过共享的三角瓶颈在两个等效扭曲八面体位点之间迁移,以及在“三帽三棱柱”多面体内部的四面体间隙跳跃迁移。这种迁移方式具有低扩散势垒和极高的锂离子扩散系数,可实现锂离子的快速传导。同时,孙学良等研究人员[52]在SmCl3框架中通过理论计算和实验表征发现一种普遍适用的界面键合及相互作用机制和离子扩散行为。通过吸附金属离子半径较小的卤化物,可以在不改变基底框架结构的情况下获得载流子锂离子,从而实现了锂离子的快速迁移。
总的来说,金属氯化物通过离子键在阴离子堆叠层之间实现了锂离子的快速迁移,同时晶体结构中的反位缺陷、杂质和晶界可能会影响离子的迁移,这是我们在材料设计和制备过程中需要考虑的问题。
5 金属氯化物固态电解质的性能优化策略
金属氯化物固态电解质的室温离子电导率(10-3S/cm)仍远低于先进硫化物固态电解质(10-2S/cm)。分子动力学模拟结果表明,基于独特hcp和ccp氯阴离子亚晶格的Li3MCl6可实现更高的室温锂离子电导率(14~29 mS/cm)[33],表明其离子电导率有进一步优化与提升的空间。现阶段,主要通过异价阳离子及卤素阴离子掺杂、结构无序化、氧阴离子掺杂及高熵氯化物等策略提升其室温离子电导率。
5.1 异价阳离子及卤素阴离子掺杂
异价金属阳离子或卤素阴离子的掺杂可以通过改变锂离子的相对含量、缺陷浓度或晶格极化率,以增强离子电导率[图8(a)]。在2020年,加拿大滑铁卢大学Park K H 等研究人员[66]通过对Li3YCl6和Li3ErCl6进行Zr4+掺杂改性,诱导了金属离子重排,使得晶体从三方结构转变为正交结构,随之产生了新的锂四面体,其占据部分位点(占据率仅为20%),并在原有的八面体位点中产生了大量的空位缺陷(25%~80%),形成了三维锂离子传输网络,使得室温离子电导率从10-4S/cm 提升至10-3S/cm;随后,韩国延世大学Yoon S J等研究人员[47]发现使用Fe3+部分取代Li2ZrCl6不仅可以缓解金属离子之间的库仑排斥力,还可以通过多面体收缩促进锂离子传输通道扩展,增强锂离子传导,为离子电导率优化提供了新的设计思路。此外,美国佐治亚理工学院的Chen Hailong 等研究人员[67]最近通过Br-取代方案获得了Li3YBrxCl3-x混合卤素固态电解质,其热压粉末的室温离子电导率高达7.2 mS/cm。相较于Li3YCl6,卤素阴离子的混合可以诱导晶体结构转变,实现四面体锂占据态与八面体锂占据态共存,随着八面体的空位缺陷浓度升高,进而触发锂离子协同扩散。此外,混合卤素阴离子还具备进一步软化晶格的作用,可促进晶界融合降低晶界阻抗,有利于实现优异的固-固接触,提升其本征及固态电池中复合正极内部的有效离子电导率。因此,离子掺杂改性通常会导致原始晶体结构畸变或转变,同时会引起亚晶格局部化学环境的变化,影响锂离子分布浓度及缺陷浓度,一定程度上可以提升金属氯化物的室温离子电导率。
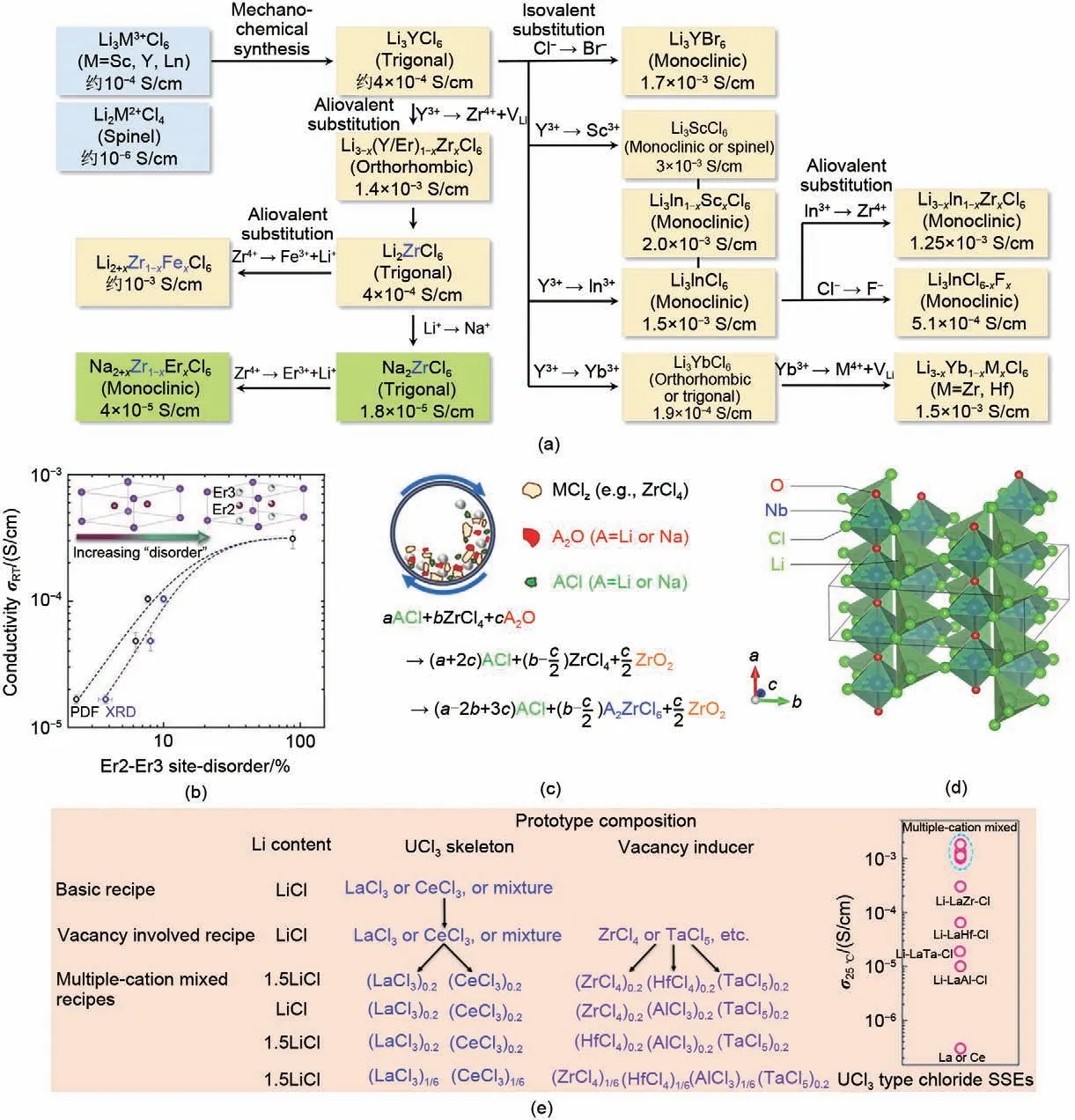
图8 金属氯化物固态电解质掺杂改性策略[46,58,68-70](a) 金属离子及卤素阴离子掺杂策略;(b) 结构无序化策略;(c)、(d) 氧阴离子掺杂策略;(e) 高熵氯化物策略Fig.8 Metal chloride solid electrolyte doping modification Strategy [46,58,68-70](a) Metal ion and halogen anion doping strategy; (b) Structure disordering strategy; (c)、(d) oxygen anion doping strategy; (e) high entropy chloride strategy
5.2 结构无序化
有序-无序结构通常会导致不同的局部亚晶格,通过改变锂离子占据位点及扩散通道,进而影响固态电解质的离子电导率。2019 年,德国吉森大学Schlem R 等研究人员[46]通过比较高能球磨和高温共熔烧结所得的Li3ErCl6的局部亚晶格信息,揭示了控制有序-无序结构对于提升离子电导率的重要作用。具体地,高能球磨法制备的Li3ErCl6中存在高度无序状态,如Er2 与Er3 占据位点置换[图8(b)],随着Er2与Er3位置无序度的增加,其八面体位点占据率会发生显著变化;且库仑斥力效应会引起锂离子无序,改变晶格中有效载流子浓度,同时晶格畸变将扩展锂离子传输通道,实现锂离子快速传导。反之,高温共熔烧结会提升结晶度,降低了无序化程度,因此导致了极低的离子电导率及相对较高的迁移活化能。所述结果表明,合成方法学对于调控有序-无序程度较为重要,基于无序化诱导效应可以显著影响局部亚晶格化学环境及其离子扩散性能[46]。
5.3 氧阴离子掺杂
近期,研究人员发现将适量的氧阴离子引入氯化物晶体骨架中,会大幅度提升所得产物的室温离子电导率。韩国延世大学Yoon Seok Jung 等研究人员[69]通过机械化学作用合成了一类氧氯化物纳米复合固体电解质ZrO2-LiCl-Li2ZrCl6[图8(c)],局部阴离子取代效应导致离子传输通道加宽和界面处局部锂离子浓度增加,在界面处形成超离子传导的纳米结构网络,使其室温离子电导率从0.4 mS/cm提升至1.3 mS/cm。此外,日本松下电器产业株式会社Yoshiaki Tanaka 等研究人员[58]发现,通过氧阴离子的引入,可以在金属氯化物晶体框架中形成共角连接的[NbO2Cl4]3-一维八面体链[图8(d)],锂离子可占据链中的四面体间隙位点。由于该链在三维空间中形成了贯通的传导网络,分子动力学研究结果表明传导路径的阴离子框架只包括一价的氯阴离子,消除了二价的氧阴离子对锂离子的束缚作用,由此实现了目前金属氯化物基固态电解质室温离子电导率的最高值,为10.4 mS/cm,接近于液态电解质传导水平。随后,加拿大西安大略大学孙学良等研究人员[71]通过氧离子介入,提出一类xLi2OTaCl5无定形金属氧氯化物固态电解质,通过构建无序且不规则排列的[Ta-Cl-O]基元,造成锂离子位点广泛扭曲,同时扩大离子迁移通道半径,由此产生的无序结构可实现高达6.6 mS/cm 的超快离子传导。
5.4 高熵氯化物
研究发现,通过采用高熵策略增加快离子导体框架的化学成分复杂性,不仅可以维持快离子导电的结构框架,还能引入额外的化学无序亚晶格,进而构建渗透连接网络促进离子快速迁移。近日,宁波东方理工大学的孙学良等研究人员[70]利用UCl3框架的丰富通道位点,采用丰富的Al、La、Ce 和Zr制备了多种具有成本效益的高离子电导率的多阳离子混合金属氯化物高熵固态电解质。基于高熵阳离子无序效应和UCl3独特的孔道结构,可以实现一系列单价离子(如Li+、Na+、Cu+、Ag+)的室温快速传输。其中锂离子的室温电导率可以提升4 个数量级,达到10-3S/cm[图8(e)]。
6 金属氯化物固态电解质的电极-电解质界面兼容性
除了固态电解质的本征离子电导率,电极-电解质界面稳定性亦是全固态锂电池性能与使用寿命的关键性限制因素。电极-电解质界面的兼容性及运行稳定性决定了全固态电池的正负极材料的选择,进而影响其质量/体积能量密度。同时,它通过影响固态电池内部的离子传导速度,决定其输出的功率密度[72-76]。与氧化物、硫化物固态电解质相似,金属氯化物固态电解质同样面临着复杂的电极-电解质界面系统。常见的电极-电解质界面问题主要体现在物理接触性、化学兼容性以及电化学稳定性三个方面[72]。尽管实验结果和理论模型已经证明,金属氯化物是一类综合性能优异的固态电解质材料,但基于金属氯化物的全固态锂电池性能仍然受到电极-电解质界面的限制,需要进一步深入研究[64]。
6.1 负极界面兼容性
金属氯化物的负极界面兼容性取决于非锂阳离子的耐还原稳定性。密度泛函理论模拟结果显示,Li3YCl6及Li3ScCl6的负极稳定电化学窗口分别是0.62 V及0.91 V[图9(a)],当其电位低于电化学稳定窗口时,氯化物电解质中的三价金属阳离子会发生还原反应,生成金属单质及氯化锂层[33,45]。因此,现有的金属氯化物固态电解质不能与锂金属负极直接匹配,阻碍了全固态锂金属电池的基础与应用研究。2021 年,德国明斯特大学Jürgen Janek 等研究人员[77]通过锂金属溅射沉积工艺,结合原位X射线光电子能谱揭示了Li3InCl6与锂金属界面之间的化学信息变化,证明了界面处锂金属诱导In3+持续发生还原反应,导致固态电解质不断降解,界面还原反应的产物无法完全阻断电子交换,会诱导电解质中过渡金属阳离子持续还原降解,故导致金属氯化物固态电解质-锂负极界面不兼容。
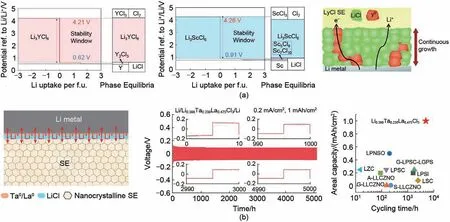
图9 金属氯化物固态电解质负极界面兼容性[33,45,53](a) Li-M-Cl型金属氯化物负极界面兼容性;(b) UCl3型金属氯化物负极界面兼容性Fig.9 Anode interface compatibility of MCSEs[33,45,53](a) anode interface compatibility of Li-M-Cl type MCSEs; (b) anode interface compatibility of UCl3 type MCSEs
目前,通常采用自限制型界面设计与合金型负极设计稳定金属氯化物负极界面。自限制型界面主要是通过引入Li6PS5Cl固态电解质层,通过产生的LiCl、Li2S 及Li3P 组分构建动力学稳定界面,阻断副反应的持续进行,从而实现界面钝化。2021年,美国威斯康星大学密尔沃基分校Qu Deyang 等研究人员[78]及加拿大西安大略大学孙学良等研究人员[36]通过引入Li6PS5Cl 电解质,诱导锂金属表面自限制型界面层的产生,构建了循环稳定的Li|Li6PS5Cl|Li3YCl6|NCM全固态锂金属电池。需要指出的是,硫化物与氯化物固态电解质界面稳定性仍有待提高,基于自限制界面层的稳定作用会随长循环而逐渐失效,其组分调控仍然需要进一步优化。同时,自限制型界面层作为非活性组分,严重限制了全固态电池的能量密度。因此,如何实现金属氯化物固态电解质与锂金属之间的热力学或动力学稳定性仍然是解决锂负极不兼容的关键科学问题。
2023 年,本课题组首次发现镧系元素具有过渡金属元素中最低的电负性,理论上具备最佳的负极耐还原性,结合所产生的电绝缘LiCl界面相形成的梯度钝化层,不仅可以阻断非锂阳离子的持续还原,还可以有效地缓解锂沉积/剥离过程中的界面应变,所组装的锂金属对称电池以0.2 mA/cm2的电流密度和1 mAh/cm2的面容量可稳定循环5000小时以上[图9(b)][53]。
此外,新型合金负极,因其高离子扩散系数、高容量及合适的工作电位,被认为是锂负极的有效替代品[76,79]。现阶段常用的合金负极包括Li-Si 合金[80]、Li-Sn 合金[81]、Li-Al 合金[82]及Li-In 合金[83]等,其工作电位分别是0.3 V、0.3 V、0.35 V、0.62 V。合金型负极不仅可以避免高活性锂诱导电解质降解,同时可以避免锂“枝晶”的产生,保证电池运行安全。然而,其高工作电位使得其能量密度无法与商业化锂离子电池媲美。
综上所述,理论与实验结果均证明Li-M-Cl 型金属氯化物固态电解质因含有易被还原的金属离子,故与锂金属负极在热力学上不稳定,在循环过程中无法生成自限制型的界面钝化层,将导致负极界面持续恶化,从而不适用于全固态锂金属电池。此外,UCl3型固态电解质的相关实验结果证明了非锂阳离子类型对于界面稳定性至关重要,通过合适的阳离子设计寻找界面稳定的氯化物固态电解质仍然需要进一步深入探究[73]。
6.2 正极界面兼容性
热力学理论分析揭示金属氯化物固态电解质-正极界面稳定性明显优于硫化物固态电解质-正极界面[图10(a)][68]。因此,金属氯化物固态电解质表现出优异的正极界面兼容性[32,51,84,85],当匹配高电压LiCoO2、LiNi1-x-yCoxMnyO2、LiNi1-x-yCoxAlyO2材料时,均可构建稳定循环的全固态锂电池。这主要归因于氯阴离子框架独特的化学性质[63,86,87]:
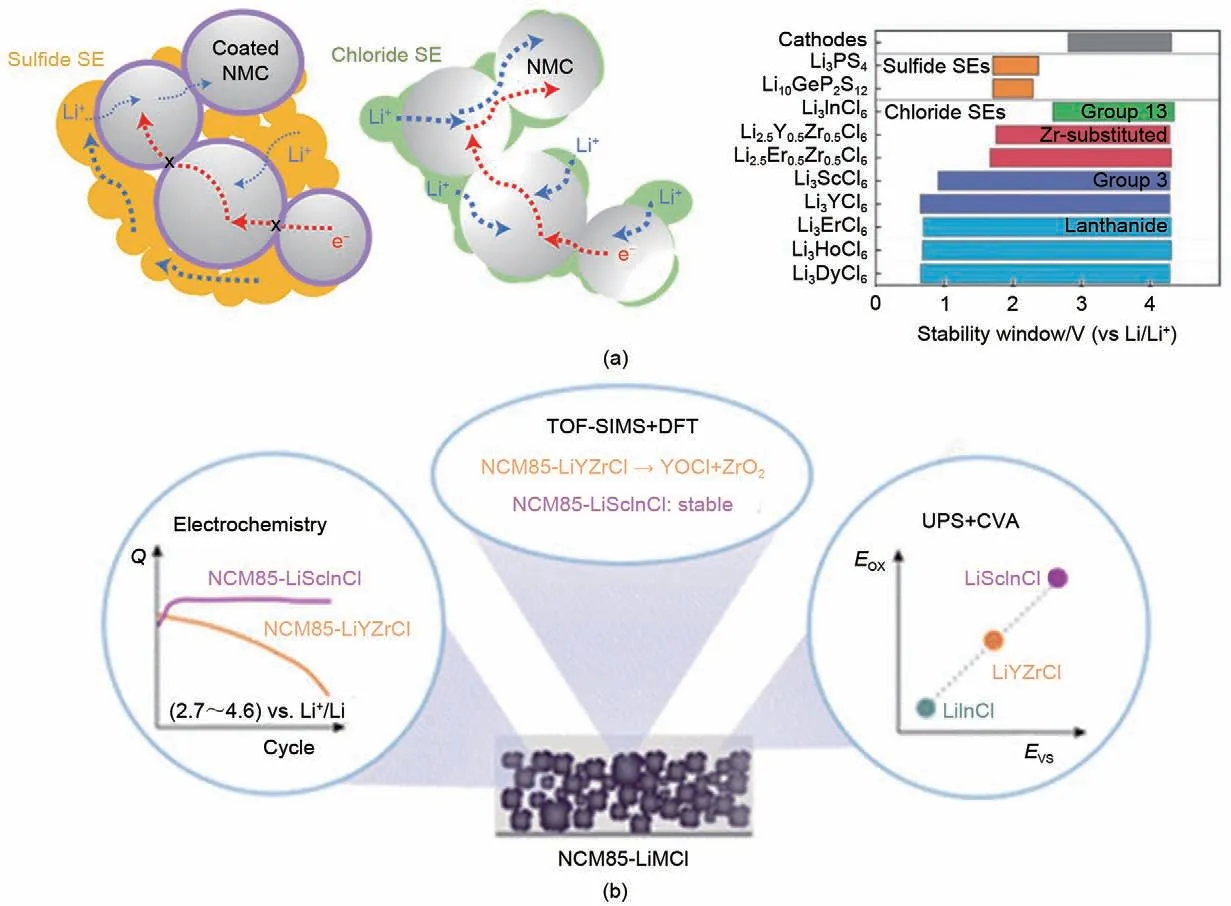
图10 金属氯化物固态电解质正极界面兼容性(a) 金属氯化物--正极界面接触性及电化学稳定性[51,89-90];(b) 金属氯化物--正极界面衰减机制Fig.10 Interface compatibility of MCSEs and cathode [51,89-90](a) MCSEs-cathode interface contact and electrochemical stability; (b) MCSEs-cathode interface decay mechanism
(1)Li—Cl离子键较长,极化率高,晶格延展性好,可缓解正极晶格充放电过程中的体积膨胀收缩,有利于构建并保持紧密接触的电极-电解质界面;
(2)Cl-电负性高(χCl=3.16),接近于O2-(χO=3.44),赋予了金属氯化物耐氧化性,高电压状态下不会氧化分解,保证了复合正极内部有效的锂离子传输网络。
基于优异的化学相容性,由金属氯化物固态电解质构建的全固态锂电池表现出优异的电化学性能[32,51],包括高库仑效率、低电极-电解质界面阻抗、长循环稳定性及高电压循环稳定性。然而,2022年加州大学圣地亚哥分校孟颖等研究人员[88]发现高电压(>4.5 V)下,Li3YCl6的氧化分解将会导致电池内阻明显增加,诱导容量迅速衰减,最终造成严重的电池失效。因此,为实现稳定的高压体系全固态锂电池,金属氯化物固态电解质-正极界面仍然需要进一步改善与优化,以减小金属氯化物氧化分解对高压正极稳定运行的影响。
由上述讨论可知,固态电解质-电极界面稳定性取决于其化学兼容性和电化学稳定性。虽然金属氯化物固态电解质具有较高的本征高电压稳定性,但为了构建更加稳定的高电压电极-电解质界面,需要重新思考正极界面工程策略,包括涂层包覆和掺杂改性在改善固态电池循环稳性中的重要作用。
需要指出的是,金属氯化物固态电解质材料体系虽然均表现出优异的热力学氧化电位,但其相应的全固态锂电池循环性能却明显不同,表明金属氯化物固态电解质-电极界面化学/电化学稳定性的决定因素不仅仅是氯阴离子亚晶格。2022年,加拿大滑铁卢大学Linda F.Nazar与德国吉森大学Jürgen Janek 等研究人员[90]采用电化学交流阻抗谱、飞行时间二次离子质谱技术(ToF-SIMS)结合第一性原理计算(DFT)对Li3InCl6、Li2.5Y0.5Zr0.5Cl6、Li2In1/3Sc1/3Cl4电解质的性能和其相关的正极界面进行系统的分析,首次提出非锂阳离子的种类在控制正极侧固态电解质界面膜组成和高电压循环稳定性中的重要作用。实验与理论结果表明,In3+和Sc3+非锂阳离子有利于金属氯化物固态电解质与充电状态的三元正极构建动力学稳定界面;而Li2.5Y0.5Zr0.5Cl6与三元正极的副反应会导致氯氧化钇副产物的积累,从而加快容量衰减;同时,伴随着热力学稳定的ZrO2的形成,高放热动力学会促进其界面副反应持续发生,最终导致高电压循环下的固态电池迅速失效[图10(b)]。
综上所述,金属氯化物固态电解质-正极界面稳定性主要取决于两方面的因素,其一是固态电解质本征的电化学稳定性;其二则是电解质-电极界面热力学稳定性及化学兼容性。此外,为了实现金属氯化物基的全固态锂电池的稳定运行,除了优化材料本征的电化学稳定性,正极界面工程也是有效策略之一,具备快速导锂能力的涂层设计可以有效阻断界面副反应,而不影响界面处的离子传导,从而解决其热力学不稳定及化学不兼容的问题。
7 金属氯化物基全固态锂电池实用化分析
2018—2023 年间,金属氯化物固态电解质基础研究领域进展迅速,已经实现了结构原始创新和关键性能突破[64,68,91]。理论与实验结果表明,金属氯化物固态电解质具有优异的电化学氧化稳定性、机械延展性及可观的离子传导性,被视为具备发展潜力的下一代固态锂电池关键材料体系。为了推进金属氯化物基的全固态锂电池实际应用进程,必须要综合考虑其基础性能、成本效益、湿度稳定性及集成电池的能量密度等各项指标。
7.1 成本效益
相较于硫化物固态电解质原料Li2S 的高昂成本,金属氯化物固态电解质组成元素的价格及地壳丰度具备产业化合成的潜力。具体而言,金属氯化物固态电解质体系中,Li2ZrCl6是目前具备高性能与成本效益的材料体系。相较于高成本、低丰度的Sc、In而言,Zr的高地壳丰度可以大幅度降低原料成本。同时,合成Li2ZrCl6前驱体的成本远低于经典的Li6PS5Cl硫化物材料[图11(a)],突破了固态电解质成本高昂的瓶颈,展现了其实际应用的潜力[64,68]。
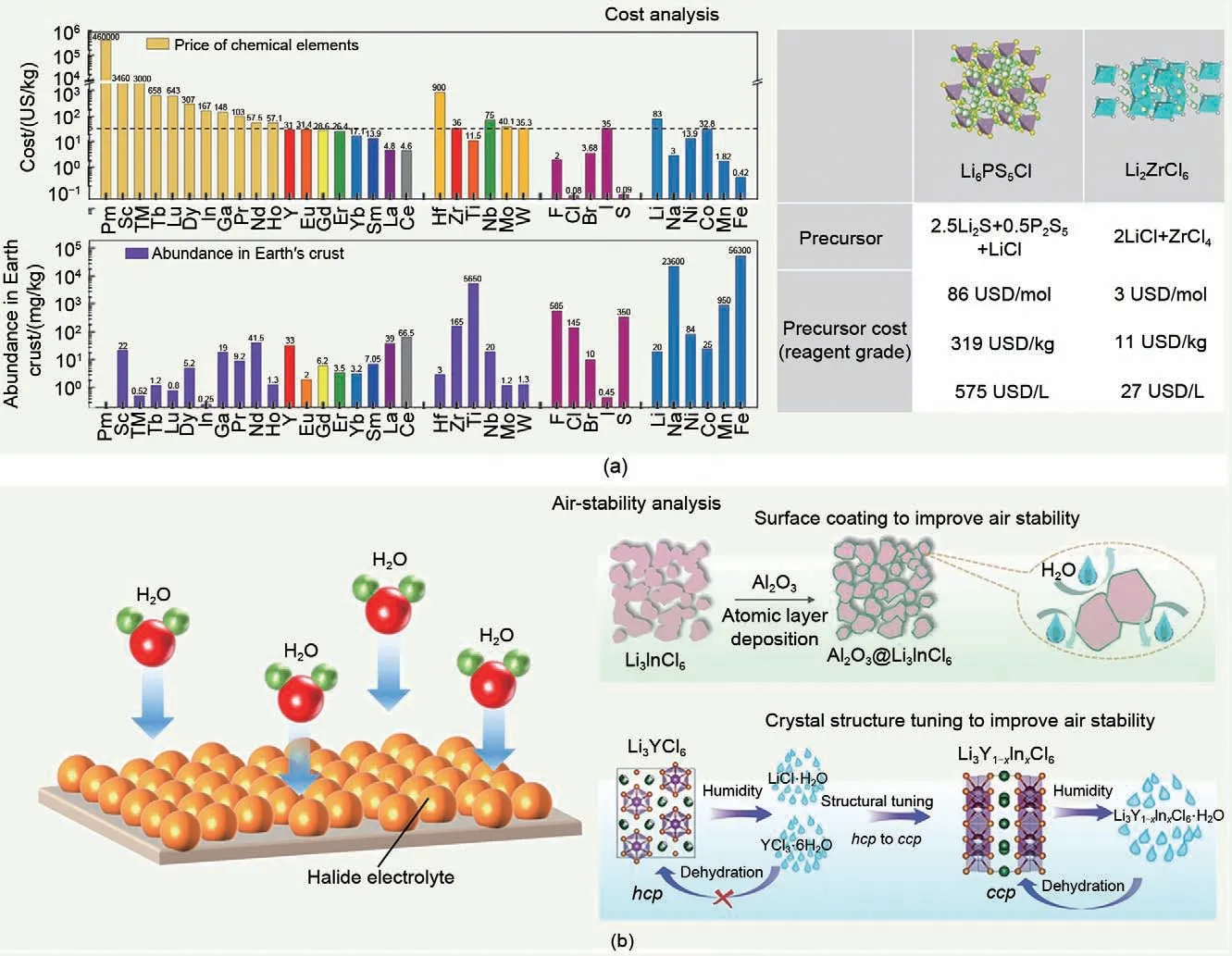
图11 (a) 金属氯化物成本效益分析[64,68];(b) 金属氯化物空气稳定性分析[64,92-93]Fig.11 (a) Cost-effectiveness analysis of metal chlorides[64,68] and (b) air stability analysis of metal chlorides[64,92-93]
7.2 湿度稳定性
金属氯化物固态电解质对湿度较为敏感[64,92-94],在水分子的持续攻击下,最终导致电解质完全水化降解[图11(b)]。湿度不稳定性对其合成、存储及全固态电池组装均提出苛刻的环境条件要求,这无疑会带来一系列的成本及造价问题。现阶段,可以通过原子层沉积在金属氯化物电解质表面引入氧化铝包覆层[92]以隔绝水分子侵蚀,提高其本征湿度稳定性。然而,氧化物作为不良锂离子导体引入电解质体系,会大幅度降低宏观离子电导率,此外,非锂阳离子(In3+)掺杂,可改变氯阴离子亚晶格构型,并通过形成稳定的水合中间体,缓解金属氯化物本征的降解,以提高其湿度稳定性。现阶段,关于如何提高氯化物固态电解质的湿度稳定性的研究有限,需要进一步对其进行结构设计及组分优化,以提高本征湿度稳定性。
7.3 高比能金属氯化物基全固态锂电池关键参数
为了赋予全固态锂电池与商业化锂离子电池媲美的能量密度[64,95],其各组件现需满足特定的设计参数及性能标准(图12)。现有的实验室规模体型模具电池因有限的面容量(约1 mAh/cm2)及极低的充放电倍率(约0.1 C),并不满足上述性能指标[32,37,45,48,60]。此外,为了进一步开发实用型全固态锂电池,必须发展合适的成膜工艺构建满足性能指标(表1)的高面容量全固态软包锂电池。基于聚合物纤丝化方法,在干燥状态下研制固态电解质复合膜以及复合正极电极片,相比于传统的湿化学方法,该方法可以很好地保持金属氯化物固态电解质的稳定性。通过采用成熟的层层叠片的方法,将复合电解质膜、复合正极电极片与锂金属负极多层叠加并封装制备全固态锂金属软包电池,构建高比能实用型全固态锂电池。

表1 高比能金属氯化物基全固态锂电池设计参数[64]Table 1 High specific energy metal chlorides based solid state lithium battery design parameters[64]
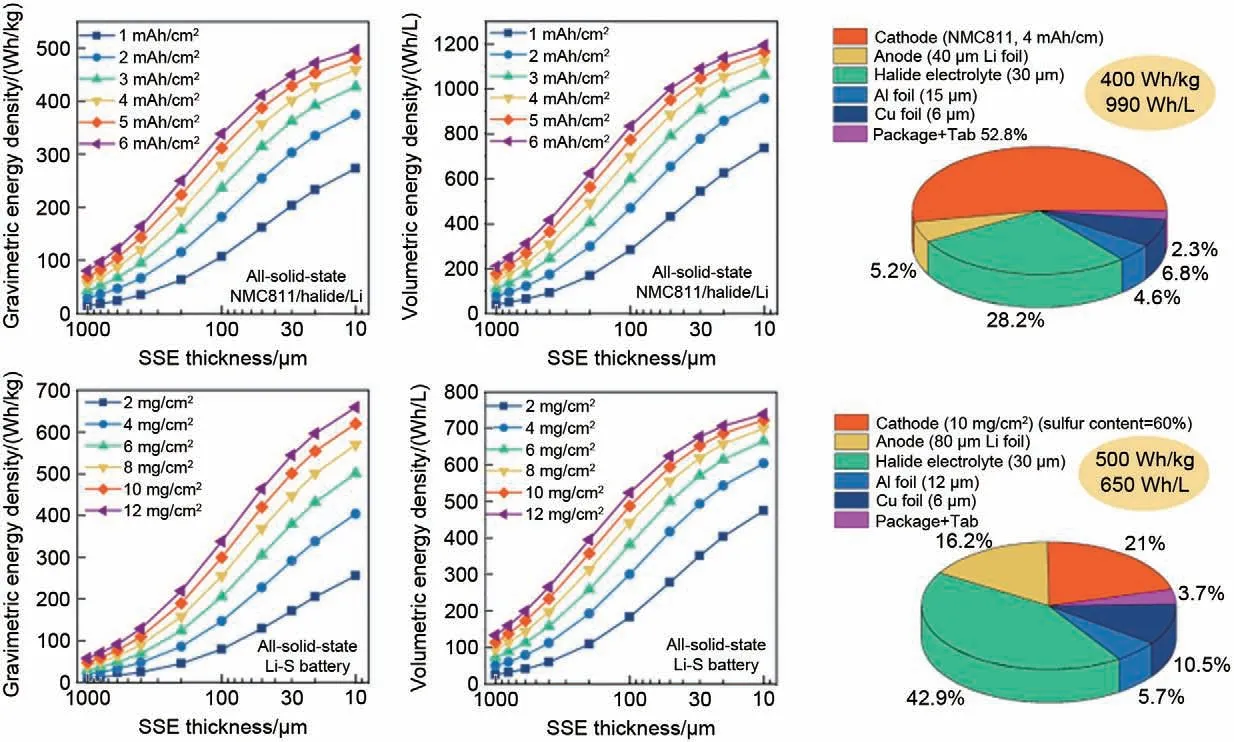
图12 高比能金属氯化物基全固态锂电池关键参数设计[64]Fig.12 Design of key parameters of high specific energy metal chloride based solid-state lithium battery[64]
综上所述,为了实现金属氯化物固态电解质的应用,仍然需要进一步提高其本征离子电导率,以实现高倍率充放电;同时提高本征离子传导性可以使得复合正极具有良好的离子渗透网络,提高正极材料的容量发挥。进一步地,锂金属负极化学/电化学不兼容性是制约金属氯化物基全固态电池产业化进程的关键问题,需进一步调控金属氯化物晶体结构并优化组分,改善锂金属负极界面热力学稳定性;或者通过发展功能界面涂层实现锂金属负极的稳定运行。
8 总结与展望
2018—2023 年间,得益于金属氯化物固态电解质材料的结构创新和性能突破,基于金属氯化物电解质的全固态锂电池亦取得了重大实质性进展。本文综述了金属氯化物固态电解质的合成方法、晶体结构、离子传导机制、性能优化策略、电极-电解质界面兼容性以及实用化可行性分析。首先,概括了目前用于金属氯化物固态电解质合成的固相及液相反应法,并讨论了合成方法的适用范围及对离子电导率和晶体结构的影响。高能球磨法多产生低结晶度或玻璃-陶瓷亚稳态材料,富含较多的本征缺陷或较高的结构无序度,有利于实现锂离子的快速传导,常用于开发新材料及初步性能验证。高温烧结法可以调控反应温度和反应速率,合成结晶度高的金属氯化物材料,可用于晶体结构解析。液相合成法目前包括水相介导合成及铵盐辅助液相合成,由于金属氯化物本身的吸湿性,其适用范围较窄。其次,通过对其晶体结构的分析,明晰了其中锂离子的传导机制,提出了一般设计原则和相应的性能优化策略。金属氯化物的晶体结构主要可以分为Li-M-Cl 密堆积型晶格及UCl3非密堆积型晶格,其锂离子迁移的基本单元均是由多面体稳定位点和过渡态位点组成,从而构建一维传输通道或三维贯通网络。通过阴阳离子掺杂、无序化晶格及高熵组分设计等策略可以进一步改善其离子电导率。最后,通过讨论电极-电解质化学及电化学兼容性,成本效应及湿度稳定性等实用化关键参数,总结了构建高比能全固态锂电池的性能参数。
然而,现阶段全固态锂电池实际能量及功率密度均低于商业化锂离子电池,限制了其在高功率型电机驱动、快速充电及大规模储能等方面的应用。为了进一步完善并推进现有的全固态电池技术,现阶段仍需关注以下基础科学问题。①开发高室温离子电导率的固态电解质:实现高功率密度全固态电池的核心在于开发高离子电导率的固态电解质材料。然而,目前金属氯化物基固态电解质的室温离子电导率仍然低于先进的硫化物固态电解质(32 mS/cm)[96]。可以通过阴离子框架设计、无序化结构优化及高熵锂亚晶格调控,明确晶体结构与离子传导性能之间的一般构效关系,进一步优化室温离子电导率。②明晰固态电池中高镍含量的层状氧化物的衰减机制:基于高镍含量的层状氧化物正极可以提供更高的比容量,进一步提升全固态电池的能量密度,但仍受到循环性和热稳定性的困扰。明确衰减机制及其与金属氯化物相互作用的机制对于提升其循环稳定性至关重要。③发展金属氯化物固态电解质干法成膜工艺:固态电解质成膜工艺是推进实验室级别模具电池向工业化级别软包电池转变的工艺基础。首先,通过开发合适的改性策略,如掺杂及包覆,提升金属氯化物的湿度稳定性的同时保持其快速的离子传导能力。随后,通过将高离子电导率固态电解质和可纤丝化的聚合物在干房中混合压延,探索其相对比例、压延压力及温度等参数获得高性能自支撑柔性固态电解质膜。构建高比能负极-固态电解质膜-高镍正极全电池体系,以实现性能优异的全固态软包锂电池。④从全固态锂电池到全固态钠电池:基于高丰度及低成本的钠资源构建高能量密度且高安全性的全固态钠电池是极具前景的下一代储能技术。全固态钠电池与全固态锂电池在许多方面有着相似的工作原理。它们都利用固态电解质在电极之间传输离子,从而实现电能的存储和释放。然而,由于钠离子的半径比锂离子大,因此在设计全固态钠电池时需要考虑更多的因素。考虑到金属氯化物的可设计性强,通过调整其化学组成和晶体结构,来优化其离子传输通道,从而适应钠离子较大的半径,实现钠离子的快速传导,并基于此研究其一般构效关系,提出金属氯化物钠离子导体设计原则及优化策略。
