纳米硅的砂磨宏量制备及其碳纤维复合负极的储锂性能研究
2024-01-26徐铖杰黄玉林林中飞雨林志铭方辰希张卫军黄志高李加新福建师范大学物理与能源学院福建省太阳能转换与储能工程技术研究中心福建福州3507
徐铖杰,黄玉林,林中飞雨,林志铭,方辰希,张卫军,黄志高,李加新(福建师范大学物理与能源学院;福建省太阳能转换与储能工程技术研究中心,福建 福州 3507)
随着新能源汽车行业的蓬勃发展,动力锂电池市场需求的急剧扩张对动力锂电池的能量密度及功率密度的要求也日益严苛。理论比容量高的硅基材料体系被认为是最有发展潜力的体系之一,更能匹配未来的市场发展[1-4]。然而,硅碳等硅基材料中的硅在锂化时体积膨胀巨大,产生的应力将导致电极材料的粉化甚至从集流体脱落,并使得活性物质不断消耗电解液,无法形成稳定的固态电解质膜(SEI膜),导致材料性能衰退;而且,硅的电导性欠佳也会导致严重的电极极化,会降低锂离子的扩散速率,限制了电池的输出功率[5-7]。研究人员常将硅纳米化处理并与碳材料进行复合,再通过复合材料的结构设计来解决上述问题。纳米硅的引入可缓解机械应变,有效地适应体积膨胀,抑制材料破裂,提高循环性能;碳材料的纳米复合可实现材料的电导调控,提高电极反应活性和面积,缩短锂离子扩散距离,提升锂离子的扩散速率,最终改善电池循环容量和倍率性能等[8-9]。
具体而言,考虑到硅体积变化及导电性较差,常见的硅基负极材料改性方法有纳米化、合金化、制备SiOx材料和硅碳复合材料等。其中,将硅纳米化是减缓硅在充放电过程中严重的体积效应最直接的办法[10-13]。Wang等[14]通过硅化钙、硫和金属钠在高压釜中的化学反应制备了平均粒径在50 nm左右的硅纳米颗粒,由该材料制备的电极在2 A/g 的电流密度下循环300 次,仍然能够保有1409 mAh/g的可逆容量。另外,通过硅碳复合的方式能够提升硅基负极材料颗粒之间的电接触、减小材料的极化,以及与电解液直接接触的概率、提升电极的库仑效率和倍率性能[15-17]。本课题组[18-19]前期通过电纺丝路径制备若干类硅碳纤维复合负极材料,特别在金属导电粒子和低电负性金属的氧化物复合修饰硅碳多孔负极的研究上取得进展,所制备的原子层ZnO修饰、金属银粒子复合的碳包纳米硅多孔碳纤维负极材料在1.8 A/g 的大电流密度下,循环1000 次后保持约920 mAh/g 的可逆容量。Hou等[20]通过还原热解法合成了一种封装在碳壳中的铜修饰硅纳米颗粒结构的复合材料(Si@Cu@CC),Si@Cu@CC电极在0.1 A/g的电流密度下循环50次后仍能保有1724.3 mAh/g 的高可逆容量,库仑效率达到了98.13%,而且在2 A/g的电流密度下也能保持1201.6 mAh/g。上述结果说明金属粒子或金属碳化物粒子与碳的协同效应能够提高电极的电导和界面兼容特性,并为锂离子和电子提供了丰富的扩散通道,最终实现硅碳复合负极性能的显著提升。
因此,选用纳米硅作为硅源,将碳作为复合材料的基体,并构建硅碳复合材料是提升硅基负极材料性能的主流途径。本工作通过优化实验工艺将太阳能电池硅废料由微米尺度砂磨至粒径约为300 nm的硅纳米颗粒。值得注意的是,将砂磨所得的纳米硅颗粒进行透射电子显微镜的原位嵌脱锂监测发现,在充放电过程中硅的横向粒径仅仅膨胀了49.7%,其体积效应已被很好抑制[21-24]。另外,将硅纳米颗粒组装扣式电池进行储锂性能分析,可媲美商化硅纳米颗粒,为进一步进行硅碳复合材料研究奠定基础。
在前一步“变废为宝”的宏量制备硅纳米颗粒的基础上,本工作进一步通过静电纺丝方法制备结构稳定的铜金属颗粒修饰的硅碳纤维复合材料(定义为Cu-Si@CNFs),并综合测试其电化学储锂性能。实验表明,得益于电纺丝特定的制备方式,所制复合材料展现出细长交错的纤维形貌,具备良好的导电性和结构稳定性;引入铜纳米颗粒修饰硅碳纤维复合材料,可提升复合材料电导及其循环过程中的结构稳定性。测试结果显示,所制备的Cu-Si@CNFs 复合材料作为锂电池负极,在经过低电流密度循环活化后,在1 A/g 的较高电流密度下循环550 次后仍可展现出765.9 mAh/g 的可逆容量,明显优于未引入铜金属颗粒修饰的纯硅碳纤维复合负极材料的储锂性能。因此,本工作为硅纳米颗粒的宏量制备工艺以及设计稳定的硅碳复合结构来调制和提升材料的储锂性具有良好的参考意义。
1 实验部分
1.1 材料的合成
1.1.1 硅纳米颗粒的砂磨制备
首先,将0.3 kg 的太阳能电池微米硅废料与0.05 kg聚乙烯吡咯烷酮(PVP)共同加入至无水乙醇中混合均匀。随后,将混合溶液投入砂磨机中以1100 r/min砂磨10 h,所得均匀且粒径约为300 nm的纳米硅浆经干燥后作为纳米硅的原料使用。
1.1.2 硅碳复合纳米纤维的制备
首先,量取10 mL的N,N-二甲基甲酰胺(DMF),并称取0.36 g的纳米砂磨硅于烧杯中磁搅均匀。接着,将溶液进行30 min 的超声处理使得纳米砂磨硅均匀分散于溶液中。随后,称取0.16 g的乙酸铜与1.28 g 的聚丙烯腈(PAN)至溶液中磁搅过夜,形成电纺丝胶体。在静电纺丝的过程中,针头与集电极保持15 cm的距离,并保持1 mL/h的进料速度。将静电纺丝所获得的纳米纤维置于真空干燥箱中以80 ℃干燥24 h,随后将其放置管式炉中以2 ℃/min的速率升温至260 ℃保持2 h。再置于氩氢混合气的气氛下以2 ℃/min的速率升温,分别在350 ℃和600 ℃的温度下保持30 min完成氧化铜的还原与复合材料的退火。最后,通过自然降温获得目标样品,标记为Cu-Si@CNFs。Si@CNFs 与Cu-Si@CNFs的制备过程相似,区别在于在溶液的制备过程中未加入乙酸铜。
1.2 材料表征
使用RIGAKU SCXmini X射线衍射仪(XRD)进行材料的晶相测定,测试扫描速率为10°/min,范围为10°~80°。使用KEITHLEY 4200A-SCS 参数分析仪对材料的电导进行分析。使用激光激发波长为780 nm 的雷尼绍共聚焦拉曼显微光谱仪获得材料的拉曼光谱。在振实密度测试实验中,将不同砂磨时间的硅材料置于量筒中,以200次每分钟的速率振动15分钟,达到振实3000次的测试标准。热重分析(TGA,耐驰STA449C)记录在10 ℃/min 的升温速率及在气流下从10 ℃到1000 ℃温度范围内进行。X 射线光电子能谱(XPS,Thermo Scientific ESCALAB250Xi)用于分析元素种类及价态。Brunauer-Emmett-Teller(BET)法测量比表面积和孔隙率。通过扫描电子显微镜(FESEM,日立,SU-8010)观察材料形貌和元素分布。
1.3 扣式电池组装和电化学测量
通过在充氩手套箱中组装的CR2025扣式电池来评估材料的电化学储锂性能。首先,将质量分数80%活性材料、10%导电炭黑和10%羧甲基纤维素钠黏结剂溶解在去离子水中,混合形成均匀的浆料,再将其涂布在铜箔集流体上。然后,将涂布好的电极在100 ℃下真空干燥12 h后并压实处理。纽扣电池由电极、锂片、隔膜(聚丙烯膜-Celgard2400)和电解液[组分为1 mol/L LiF6PO4(DC∶EMC∶DEC=1∶1∶1,体积比)]封装而成。其中,每个电极的负载密度为(1.1±0.2) mg/cm2。将封装好的纽扣电池在LAND CT2001 仪器上进行充电和放电测试,并在CHI760E 电化学工作站上进行不同扫描速率下的循环伏安法(CV)测试和频率范围为100 kHz至100 MHz的电化学交流阻抗测试。
2 结果与讨论
图1展示了以太阳能电池微米硅废料为原料并通过砂磨制备硅纳米颗粒的示意图。在砂磨机的内部,大量的白色锆珠通过碰撞、剪切力等作用将微米尺度的硅废料砂磨至纳米尺度。其中,液相溶剂为无水乙二醇和PVP 可以避免硅的氧化,而且PVP 作为分散剂使得硅能够在砂磨过程中均匀分散,抑制纳米硅团聚的现象发生。
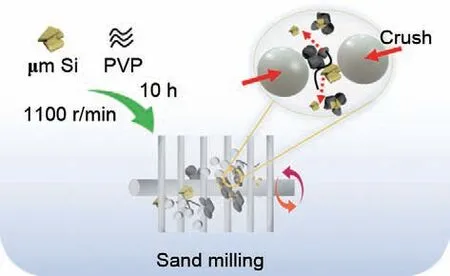
图1 纳米砂磨硅的制备示意图Fig.1 Schematic representation of the preparation of nano-silicon via sand milling
为了考察微米硅废料的最佳砂磨工艺,图2(a)~(j)给出了不同砂磨时间制备的硅纳米颗粒的扫描电镜图像,并对比微米硅在砂磨前后的形貌变化。图2(a)~(b)展示了未经过砂磨处理的微米硅废料的形貌图,可知硅原料的粒径在5 μm 以下,且不规则的大颗粒表面存在着1~3 μm 的小颗粒。如图2(c)~(d)所示,经过4小时的砂磨,部分微米硅的粒径已降至500 nm 左右,但大颗粒仍然广泛存在,无法满足进一步应用的需求。如图2(e)所示,通过8小时的砂磨,大部分的微米硅已经被砂磨至数百纳米的粒径,显示出砂磨处理是一种有效的宏量制备纳米硅颗粒的手段。而从图2(f)中可以观察到,依然有小部分的大颗粒微米硅存在,这会导致硅在充放电过程中发生严重的体积效应,造成电极材料发生严重的粉化甚至脱落[25]。进一步地,如图2(g)~(h)所示,在10 小时的充分砂磨之后,绝大部分的微米级硅颗粒已被砂磨至300 nm 左右的纳米硅颗粒,达到符合实际应用的需求。不仅如此,如图2(i)~(j)所示,在12小时的砂磨之后,材料的形貌基本与砂磨10 小时的材料无异,说明将太阳能电池微米硅废料与分散剂混合于无水乙醇中,投入砂磨机砂磨10 小时是最佳方案。同时,从图2(k)~(o)给出的对应的粒径分布柱状图可以清楚看出上述硅颗粒的粒径大小随砂磨时间延长的变化情况。值得注意的是,在本小组先前的研究工作中[21],将砂磨后的纳米硅置于透射电镜下进行嵌脱锂的原位观测表明,硅纳米颗粒在第一次锂化过程仅仅发生了49.7%的横向尺寸膨胀,而且显示出媲美市售纳米硅的膨胀和储锂性能。同时,从图2(p)~(t)所示的太阳能电池微米硅废料经不同砂磨时间后的硅浆照片可看出,未经砂磨的硅浆呈灰褐色,且硅颗粒的大小与分布不均匀。而随着砂磨的进行,硅颗粒逐渐纳米化,同时硅浆的颜色随之变化,逐渐变成了黄褐色。因此,也可以根据硅浆的颜色变化来初步判断微米硅的砂磨纳米化处理工艺,可为硅纳米处理提供直观的判断经验。
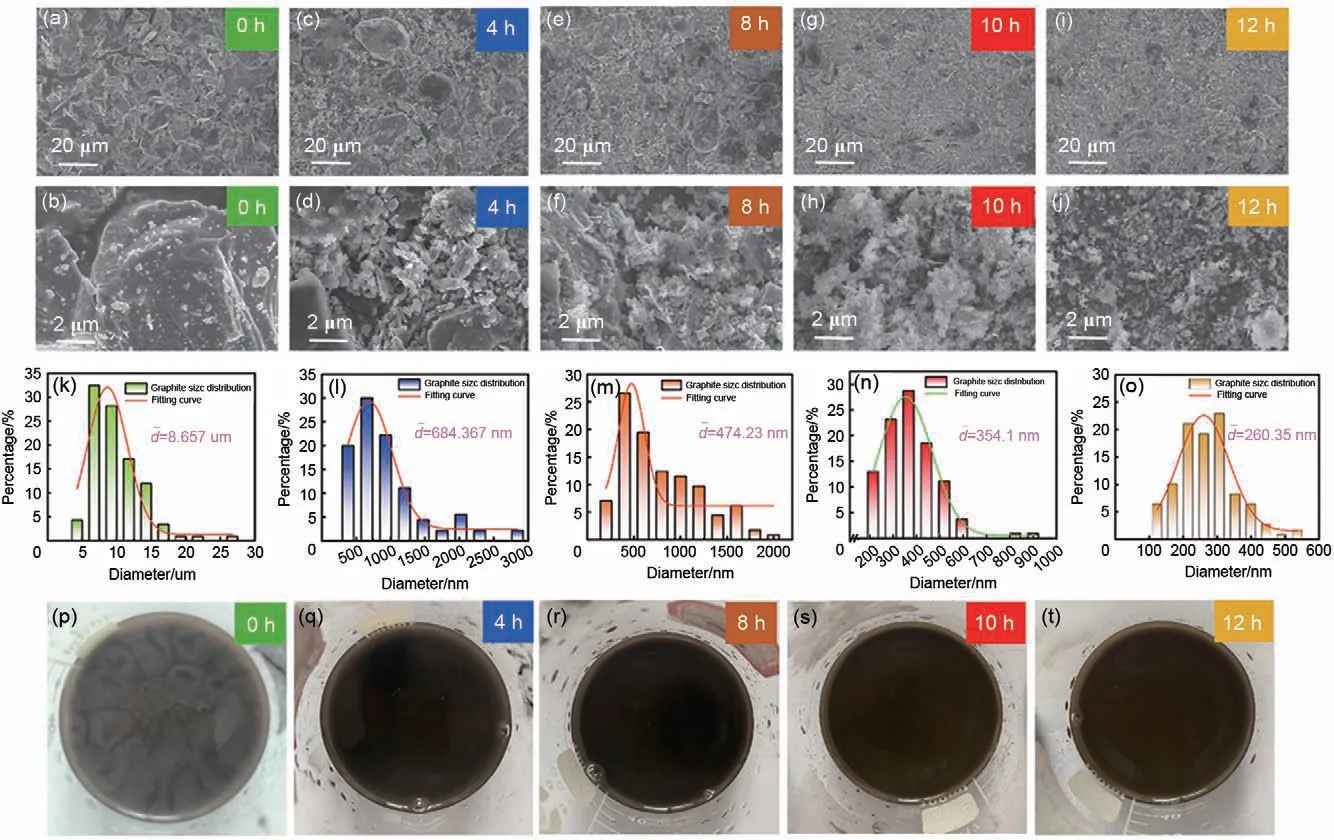
图2 微米硅废料在经历不同砂磨时间后的 (a)~(j) 扫描电镜图,(k)~(o) 对应的粒径分布柱状图及 (p)~(t) 对应的硅浆照片Fig.2 (a)—(j)Scanning electron micrographs of micron-sized silicon waste after varying durations of sand milling, (k)—(o) bar graph of the corresponding particle size distribution (p)—(t) and corresponding photographs of the silicon slurry
为进一步确认砂磨纳米硅的晶体结构,图3(a)给出微米硅废料不同砂磨时间后的X 射线衍射图。从图看出,不同的硅样品均在2θ=28.6°、47.3°、56.3°、69.3°、76.5°有明显的衍射峰,分别对应硅的(111)、(220)、(311)、(400)、(331)晶面。如图3(b)所示,通过傅里叶红外谱分析砂磨过程产生官能团。与未经过砂磨的样品相比,随着砂磨时间的增加,在810 cm-1与890 cm-1的位置观察到逐渐增强的Si—OH键信号,且在1225 cm-1和1632 cm-1的位置观察到逐渐明显的宽峰,分别对应于Si—O—C键与C—C、C=O键,这归因于硅在砂磨过程中与分散剂中少量的碳逐渐生成稳定的强健合[26-27]。相对于简单的物理接触,存在化学键有利于提高电极材料的结构稳定性与电导率[27]。图3(c)展示了不同砂磨时间样品的Raman 光谱图,在282.8 cm-1、512 cm-1、925.9 cm-1的位置分别观察到明显的宽峰,分别对应着硅的第二声子模式、晶体硅的Si—Si 键、非晶SiOx中的Si—O 键[28-29]。其中,硅由于在空气中会发生氧化形成SiOx。因此,在砂磨的作用下,少量SiOx氧化层随砂磨时间的增加而进一步形成。图3(d)给出微米硅废料不同砂磨时间样品的比表面积统计图。随着砂磨时间的增加,样品的比表面积逐渐增加,表明样品粒径尺寸的减小。值得注意的是,砂磨10小时与12小时的样品比表面积相差不大,说明砂磨10 小时是优选方案。
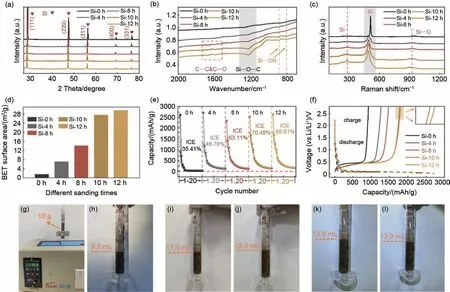
图3 不同砂磨时间硅材料的 (a) XRD图,(b) 傅里叶红外谱图,(c) Raman光谱图,(d) 比表面积对比图,(e) 前20次循环结果,(f) 首次充放电曲线以及 (g)~(l) 振实密度测试图Fig.3 Silicon materials of different sanding time: (a) X-ray diffraction patterns, (b) Fourier-transform infrared spectra, (c) Raman spectroscopy results, (d) comparative surface area values, (e) cycling results of the initial twenty cycles, (f) initial charge-discharge curves, and (g)—(l) tap density evaluation results
为了分析砂磨纳米硅在实际应用的可行性,图3(e)~(f)展示了不同的硅纳米材料的储锂性能。其中,从图3(e)展示的前20次循环充放电曲线可知,硅材料的首次放电比容量均在2300~2700 mAh/g;且随着砂磨时间的增加,硅纳米材料的循环稳定性及首次库仑效率都略有提升。而且,图3(f)的首次充放电曲线直观地说明了随着砂磨的进行,由于硅颗粒进一步被砂磨至纳米尺度,带来的是充放电过程中体积效应的减缓,砂磨10小时与12小时的样品展现出提升的性能。此外,如图3(g)~(l)所示,随着砂磨时间的增加,硅材料的振实密度逐渐降低,而砂磨10小时与12小时的样品的振实密度较为接近。这归因于随着砂磨的进行,材料逐渐纳米化,比表面积逐渐增加,振实密度由1.05 g/cm 降低至0.72 g/cm。而经过10小时的砂磨,足以将微米硅废料砂磨至约300 nm尺度,因此砂磨10小时与12 小时的样品在各项物化表征中均展现出相近的数值。综上,纳米化有助于减缓硅在充放电过程中的体积膨胀,提供更多可能的电化学活性位点[30],是一种直接有效的改善硅基负极材料性能的手段。可见,微米硅废料的砂磨处理实现硅纳米颗粒的宏量制备能够展现一定的应用潜力。
如图4(a)所示,Si@CNFs 与Cu-Si@CNFs 复合材料在28.4°、47.3°、56.1°、69.1°、76.3°位置的衍射峰分别对应于硅的(111)、(220)、(311)、(400)、 (311) 晶 面(JCPDS 75-0589); 而Cu-Si@CNFs 复合材料在43.3°、50.4°、74.1°的位置存在衍射峰,对应着铜的(111)、(200)、(220)晶面(JCPDS 85-1326)。此外,较为宽化的衍射峰表明复合材料存在Si和Cu纳米颗粒。为了探究复合材料中各个组分的含量,图4(b)给出复合材料的热重分析结果。在室温100 ℃,复合材料都发生了少量的失重,这归因于材料中微量水分的蒸发[31];在100~350 ℃,Si@CNFs 的失重略快于Cu-Si@CNFs,且随后出现平台,这是由于在空气中Cu-Si@CNFs复合材料的Cu 纳米颗粒完全氧化生成CuO[32];而在350~600 ℃,由于碳纳米纤维的燃烧,使得复合材料的重量发生了明显的下降。从图中看出,Si@CNFs中硅的含量为36.33%;而Cu-Si@CNFs完全热解后的重量42.81%来源于Si与CuO,根据投料比计算得知Si 的含量约为36.5%,CuO 的含量约为6.3%。因此,通过计算得到Cu-Si@CNFs中Cu的含量为5.1%。最后,随着温度由600 ℃继续升高,硅将在高温下氧化形成SiOx,导致复合材料的重量上升。Si@CNFs 与Cu-Si@CNFs 的拉曼光谱图如图4(c)所示,在505 cm-1、912 cm-1、1388 cm-1及1588 cm-1的位置分别出现了明显的特征峰。其中,在505 cm-1和912 cm-1的特征峰对应着Si的存在;而在1388 cm-1和1588 cm-1分别对应着代表碳中的sp3缺陷的D峰以及代表sp2杂化石墨化碳原子的E2g振动的G峰,D峰和G峰的峰强度比ID/IG值越高,则缺陷程度越高,石墨化程度越低[15]。通过峰强比值的计算,Si@CNFs 复合材料的ID/IG值为1.15,说明由PAN热解形成的碳纳米纤维主要以无序碳的形式存在,而Cu-Si@CNFs 复合材料的ID/IG值与其相差无几,说明微量铜纳米颗粒的引入对复合材料的石墨化程度未有明显影响。
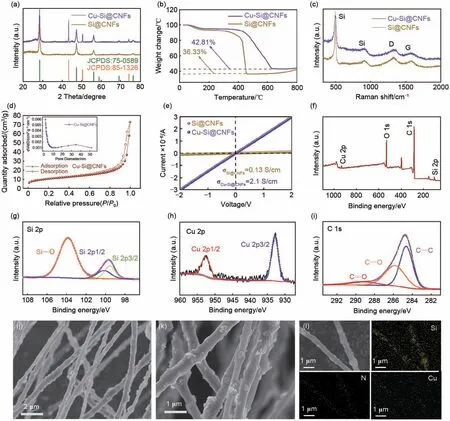
图4 Si@CNFs与Cu-Si@CNFs复合材料的 (a) XRD图,(b) TGA热重曲线及 (c) Raman光谱图;Cu-Si@CNFs复合材料的 (d) 氮气吸脱附曲线和孔径分布图,(e) I-V曲线结果以及 (f)~(i) XPS光谱图;(j)~(k) Cu-Si@CNFs复合材料不同倍数下的SEM图;(l) Cu-Si@CNFs复合材料的EDS元素分布图Fig.4 Si@CNFs juxtaposed with Cu-Si@CNFs composite of (a) X-ray diffraction patterns,(b) thermogravimetric analysis curves, and (c) Raman spectroscopy results, (d) nitrogen adsorption-desorption isotherms and pore size distribution diagrams, (e) I-V curve results, (f)—(i) X-ray photoelectron spectroscopy spectra, (j)—(k) SEM micrographs at various magnifications, and (l) EDS elemental mapping results
此外,由图4(d)的Cu-Si@CNFs 复合材料的氮气吸脱附曲线与孔径分布图可知,复合材料存在着微孔与介孔结构,且其比表面积为36.2 m2/g 和平均孔径为12.4 nm。Cu-Si@CNFs具有丰富的介孔与较大的比表面积,保证了具备较快的电解液浸润速度,可为锂离子的脱嵌提供丰富的电化学活性位点与扩散通道[33]。为了探究Cu-Si@CNFs的表面化学成分,对复合材料进行了XPS 光谱测试。同时,由图4(e)给出的两者样品的I-V曲线分析结果可知,铜纳米颗粒修饰后的Cu-Si@CNFs 的电导率为2.1 S/cm,明显高于未修饰的Si@CNFs 样品,这将为提升电极电导、降低极化及增强嵌脱锂动力学起到促进作用。如图4(f)所示,XPS全谱显示了Si 2p、Cu 2p 和C 1s 的峰,表明在Cu-Si@CNFs 材 料中存在Si、Cu 和C 元素。Si 2p 的XPS谱图主要由99.5、100.2和103.8 eV处的3个峰组成,如图4(g)所示,分别对应Si的2p3/2轨道和2p1/2轨道以及由于硅纳米颗粒表面发生氧化所生成的Si—O 键[32]。由图4(h)可知,Cu 2p 的XPS谱图主要由932.8、952.7 eV 处的两个峰组成,分别对应于铜单质的2p3/2 轨道和2p1/2 轨道[34]。如图4(i)所示,C 1s 的XPS 谱图在284.8、286.0 和289.3 eV的位置存在明显的3个峰,分别对应C—C、C—O、C=O键的结合。通过XRD与XPS的表征分析,有力地佐证了Cu纳米颗粒在硅碳复合纳米纤维中的稳定存在。从图4(j)~(k)看出,Cu-Si@CNFs复合材料整体呈现径宽在200~600 nm 的纤维形态,且硅纳米颗粒均匀分布并嵌入在纤维的表面和内部。同时,图4(l)进一步给出Si、N、Cu三个元素信号均匀地显示在纤维上且重叠良好,说明Cu 与Si在纳米纤维上均匀分布,其中N元素来源于PAN的碳化[35]。可预期,在细长交错分布的纤维主体结构上引入铜纳米颗粒可进一步促进材料导电性的提升,使得复合材料呈现出高导电的三维网格结构,将改善硅基负极材料导电性,同时有助于Cu-Si@CNFs复合材料循环稳定性与倍率性能的增强[36]。
为了研究电极的电化学动力学行为,对Si@CNFs与Cu-Si@CNFs的电极在充放循环150次前后进行了交流阻抗(EIS)测试,如图5(a)、(b)所示。EIS图主要由高、中频区的半圆,连同低频区的斜线组成。它们分别代表了欧姆电阻、锂离子经过循环后产生的SEI 膜的扩散电阻、电解质-电极界面的电荷转移电阻与电极与电解液由于浓度差而引起的浓差极化阻抗Warburg 阻抗[37]。从图5(c)看出,与Si@CNFs 电极相比,Cu-Si@CNFs 电极在循环后有着更低的扩散电阻RSEI和电荷转移电阻Rct,说明Cu 纳米粒子的引入一定程度上提升了复合材料的电导能力,降低极化并改善界面兼容特性,对电极的嵌脱锂性能的提升起到促进作用。从图5(d)给出的Cu-Si@CNFs电极的循环伏安曲线看出,电极在首次放电曲线中的0.5 V处出现还原峰,并在随后的曲线中消失,这可能是由于产生了稳定的SEI薄膜;而在大约0.18 V的位置,新的峰值出现在随后的放电曲线中,这是由于非晶态的硅转化为LixSi;在充电曲线中,可以发现在大约0.26 V和0.51 V处分别出现了两个峰值,表明LixSi合金逐渐转变为无定形的Si[38];在随后的循环过程中,曲线中峰的位置均与第一条曲线的峰重叠,显示出良好的循环稳定性;峰面积随着循环次数的增加而上升,这可归因于循环过程中材料活性的激活。
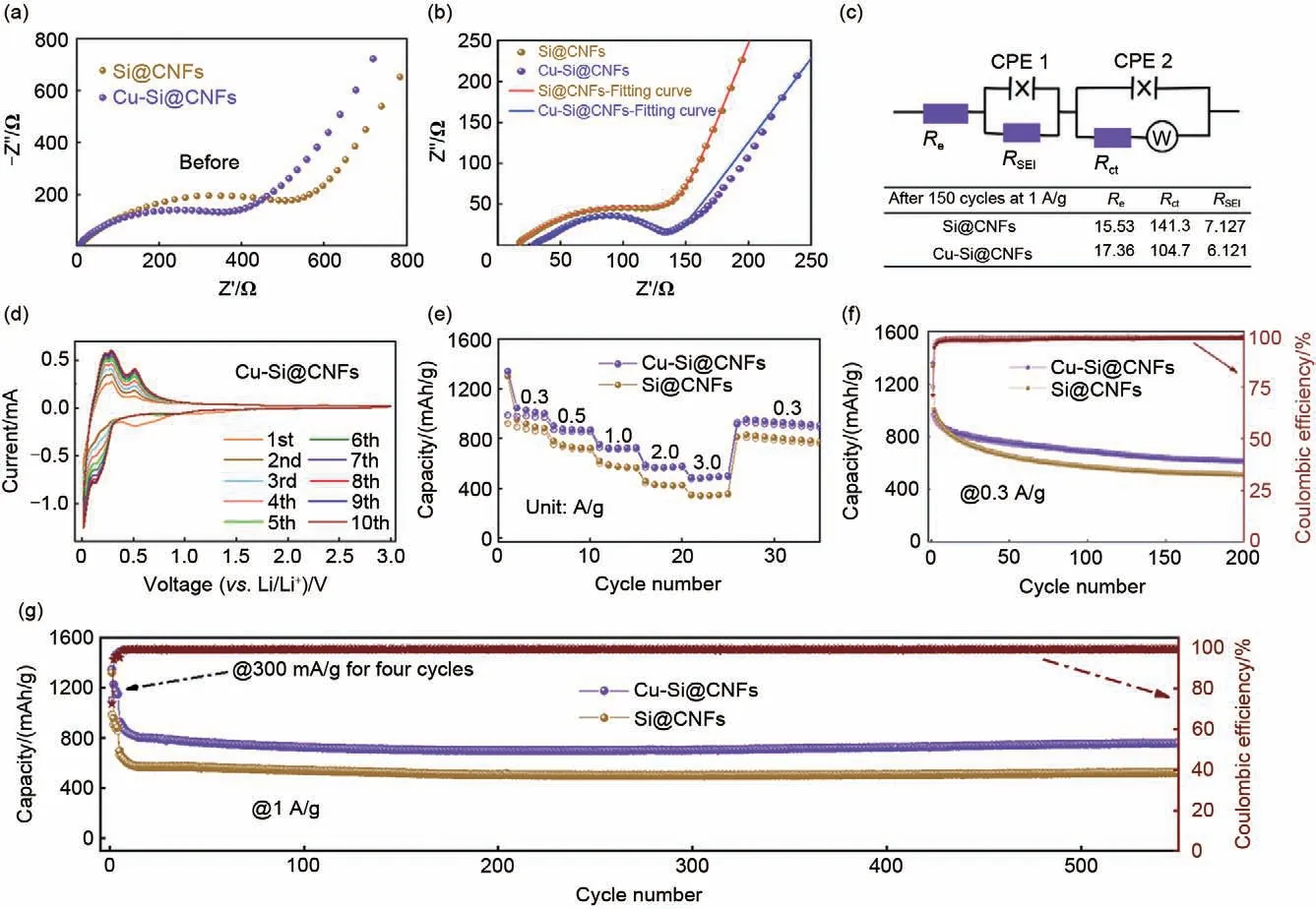
图5 (a)~(b) Si@CNFs与Cu-Si@CNFs电极循环150次前后的交流阻抗图;(c) 电极循环后交流阻抗谱的等效电路及阻抗拟合数值;(d) Cu-Si@CNFs电极在0.5 mV/s的扫速下的循环伏安曲线;Si@CNFs与Cu-Si@CNFs电极 (e) 在不同电流密度下的倍率性能曲线,(f) 在0.3 A/g电流密度下的循环曲线及 (g) 在1 A/g电流密度下的循环性能Fig.5 Si@CNFs and Cu-Si@CNFs electrodes of (a)—(b) alternating current impedance spectra before and after 150 cycles, (c) the corresponding equivalent circuit and impedance fitting value, (d) cyclic voltammetry curves at a scan rate of 0.5 mV/s, (e) rate capability curves at varied current densities, (f) cycling curves at a current density of 0.3 A/g, and (g) cycling performance at a current density of 1 A/g
图5(e)展示了Si@CNFs 与Cu-Si@CNFs 电极在0.3~3 A/g不同电流密度下的倍率性能。如图所示,随着电流密度的增加,电极的容量逐渐下降。当电流密度增加到3.0 A/g 时,Si@CNFs 与Cu-Si@CNFs 电极的容量分别下降到379.5 mAh/g 和518.5 mAh/g。值得注意的是,在每个电流密度下,Cu-Si@CNFs 电极的容量都大于Si@CNFs电极。而当电流密度由3.0 A/g 降低到0.3 A/g 时,Cu-Si@CNFs 电极保有954.2 mAh/g 的可逆容量,而Si@CNFs 电极的容量仅仅只有795.6 mAh/g。材料倍率性能的优劣与电导能力息息相关,由此说明由于Cu 纳米粒子的引入对复合材料的电导增强机制,使得Cu-Si@CNFs 电极展现出更优异的倍率性能,这与EIS阻抗所提供的结论互相印证。同时,图5(f)展示了Si@CNFs 与Cu-Si@CNFs 电极在300 mA/g 的电流密度下的循环性能曲线。在200次的循环充放电过程中,Cu-Si@CNFs电极始终相比Si@CNFs 电极有着更高的容量。在循环200 次后,Si@CNFs 与Cu-Si@CNFs 电极分别保有513.3 mAh/g 和620.8 mAh/g 的可逆容量。进一步地,为了检验电极在高电流密度下的长循环性能,由图5(g)所示,即使在1 A/g 的高电流密度下循环550 次,Si@CNFs 与Cu-Si@CNFs 电极的可逆容量仍分别保持526.9 mAh/g 和765.9 mAh/g。上述结论说明所制备的硅碳复合材料能够以纳米碳纤维作为基体,具备良好的导电性与结构稳定性,使得有效抑制碳纳米纤维中的Si NPs 在充放电过程发生的体积效应,保障了电极材料的循环稳定性[39]。不仅如此,Cu-Si@CNFs 相 比Si@CNFs 电极展示出更高的容量和更好的稳定性,究其原因在于Cu 纳米颗粒的引入进一步改善了材料的电导,降低了充放电过程中的极化,使得Cu-Si@CNFs电极展现出优异的循环性能。
不同电极的循环性能与其材料在循环中的结构稳定性等有很大的关联,为了分析铜纳米粒子引入的影响,图6 对比了Si@CNFs 与Cu-Si@CNFs 两类电极循环前后的极片形貌和界面厚度变化情况。图6(a)~(d)展示了两种电极循环150次前后的极片表面形貌图,两个电极材料在循环前均保持着较为完整且光滑的表面形貌。而循环后,由于硅在充放电过程中严重的体积效应,导致Si@CNFs 电极材料产生大量的裂纹、空隙和部分脱落,严重破坏电极的完整性;相比之下,Cu-Si@CNFs电极表面形态基本保持完整,只有较少的裂纹存在,验证了电极保持良好的完整性从而使得Cu-Si@CNFs电极展现出更优异的循环稳定性[40]。另外,从图6(e)~(h)及(i)~(j)极片界面的厚度变化分布图给出的两类极片的横截面的厚度比较看出,循环后的Si@CNFs电极的膨胀比约为46.3% [即(22.33-15.26)/15.26],而Cu-Si@CNFs 电极的膨胀比仅约为21.2% [即(20.22-16.69)/16.69]。两种电极的膨胀率相对较低,原因在于纳米纤维具有交错相连的网格结构,能够有效减缓复合材料的纵向体积膨胀,更重要的是,材料中的纳米硅颗粒均匀地嵌入纤维的表面和内部,且自身的膨胀程度相比于微米硅已大大减小,更有利于电极的长循环性能。
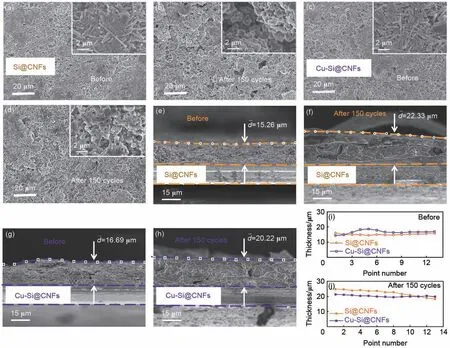
图6 (a)~(d) Si@CNFs与Cu-Si@CNFs复合材料循环150次前后的极片表面形貌图,(e)~(h) 极片截面厚度变化图及(i)~(j) 极片界面的厚度变化分布图Fig.6 (a)—(d) Evolution of electrode surface morphology for Si@CNFs and Cu-Si@CNFs composite before and after 150 cycles, (e)—(h) cross-sectional views of the electrode morphology and (i)—(j) distribution map of the electrode thickness changes
3 结 论
本工作首先以太阳能微米硅废料为原料,通过砂磨工艺的优化,可宏量制备出约300 nm 粒径的纳米硅颗粒,性能媲美市售纳米硅,实现“变废为宝”与“降本增效”的双重效用;再以宏量自制纳米硅为原料,通过电纺丝方法制备了铜纳米颗粒修饰的硅碳复合材料,并研究铜颗粒修饰硅碳复合负极材料电化学储锂性能。对制备的Si@CNFs 与Cu-Si@CNFs 复合材料进行电化学性能测试,在0.3 A/g 的低电流密度下循环200 次后分别保持513.3 mAh/g和620.8 mAh/g的可逆容量。为了探究其高电流密度下的长循环稳定性,将电极材料在1 A/g的电流密度下循环550次,分别可保持526.9 mAh/g和765.9 mAh/g 的可逆容量。将Si@CNFs 与Cu-Si@CNFs 电极在0.3~3 A/g 不同电流密度下进行倍率性能测试,Cu-Si@CNFs电极依旧有着更佳的表现。材料表征及电化学分析等表明,铜纳米颗粒的修饰能够提升复合材料的电导性,使得Cu-Si@CNFs 比Si@CNFs 展现出更好的循环稳定性与倍率性能。
