基于磁性测试揭示CoO储锂机理
2024-01-26徐熙祥阮明岳
徐熙祥,赵 越,阮明岳,李 强
(1青岛大学物理科学学院,山东 青岛 266071;2威海创新研究院,山东 威海 264299)
在当今迅速发展的社会中,能源的可持续性供应成为至关重要的问题[1]。电化学储能因其可以作为“桥梁”弥合风能、水能、太阳能等可再生能源发电端与电力分配、消费端之间的缺口而备受关注[2-3]。而精准检测电极材料中的杂质相对电化学储能系统有着极为重要的意义[4],因为杂质相的存在不仅影响储能系统的性能与效率[5],还关乎储能系统的安全性及可靠性[6]。同时,界面储能的研究与创新也对电化学储能系统发展至关重要[7],其重要性一方面在于能提升储能系统性能及容量[8],另一方面在于对其深入理解有望推动电化学储能技术革命性发展[9]。然而,无论是微量杂质相监测,还是如固体电解质界面(solid electrolyte interphase,SEI)膜等重要的界面储能机制探测,均缺乏直观、有效的表征手段[10-12]。
由于储能材料中富含过渡金属元素,材料物相、晶体结构、元素价态、电子能带、电化学性质与其磁学性质相互关联,因此磁学测试可以揭示材料中局域结构变化、元素氧化状态、杂质相及电化学机理等信息[13-15]。磁学检测技术作为一种高效、无伤、可靠的手段在过去的一些工作中已经成功应用于微量杂质相检测及界面储能机理探测中。如Ait等[16-17]利用变温磁化曲线(magnetic hysteresis, MH)测试成功探测到LiFePO4中含量仅0.7 ppm(百万分之一)超顺磁Fe2O3杂质相,并通过表面碳包覆工艺成功消除杂质相,极大提升主体材料性能;Chen等[18]则通过零场冷/场冷(zero-field-cooled/fieldcooled,ZFC/FC)测试观察到LiFePO4磁化强度在215 K附近显著上升,从而探测到微量铁磁性杂质Fe2P;Li 等[19]通过原位磁性测试发现Fe3O4放电至低电位区间磁性信号异常下降,进而基于磁学分析发掘出全新储能机理—界面空间电荷效应,从而解决困扰学界多年的“额外容量问题”。
CoO 作为一种重要材料,其在电化学储能[20]、催化[21]、陶瓷[22]等领域均有重要应用,然而其生产过程中往往无法避免地引入Co 单质杂质相[23],同时其界面储能过程也需进一步澄清[24]。基于此,我们设计实验利用磁学测试监测其杂质相及界面储能。首先,将CoO 放入H2/Ar 进行微量还原Co,制备不同比例还原样品。然后通过变温M-H测试以及ZFC/FC 测试对比传统表征手段X 射线衍射(X-ray diffraction, XRD)、X 射线光电子能谱(X-ray photoelectron spectroscopy, XPS)及高分辨透射电镜(high resolution transmission electron microscope,HRTEM)来证明磁学探测微量杂质相的可靠性以及精准性。磁学测试分别检测出CoO/Co@20min 内0.66%的Co单质杂质相,以及CoO/Co@40min内2.27%的Co 单质杂质相。同时,我们利用先进的原位实时磁学探测技术,探测到CoO 在低电压区间里复杂的空间电荷与SEI膜两种不同类型的界面储能机制,成功解释了CoO 远超理论容量的额外容量来源。本工作旨在探讨基于磁学方法如何精确检测杂质相并监测复杂的界面储能过程,从而推动储能科学领域的前沿研究并促进可持续能源的发展。
1 实验部分
1.1 CoO及CoO/Co材料制备
首先将2.49 g 四水合乙酸钴溶解于300 mL 去离子水中,搅拌10 min 使之充分混合均匀形成红色透明溶液。而后在溶液中滴入5 mL水合联氨(质量分数80%)室温下连续搅拌24 h 形成粉色悬浊溶液。再将该溶液静置12 h使前驱体充分沉淀,利用去离子水反复洗涤,沉淀物在60 ℃ 真空干燥12 h获得蔷薇色前驱体Co(OH)2。最后将前驱体在氩气中450 ℃ 退火2 h得到CoO。
所获CoO 在流动H2/Ar(体积比,5%/95%)气氛下250 ℃分别还原20 min 及40 min 制备不同比例的CoO/Co,分别记作CoO/Co@20min,CoO/Co@40min。所有材料制备煅烧过程升降温速率均为5℃/min。
1.2 材料表征
采用X 射线衍射仪(X-ray diffraction, XRD,Rigaku SmartLab SE,Cu Kα)对所合成样品进行物相分析。采用X射线光电子能谱仪(X-ray photoelectron spectroscopy,XPS,Thermo ESCALAB 250Ⅺ)
对所合成样品元素价态分析。采用高分辨透射电子显微镜 (high resolution transmission electron microscope, HRTEM, FEI Tecnai G2 F20),对样品形貌、样品精细结构进行分析。
1.3 样品电极制备及电化学测试
首先将活性物质、导电炭黑(Super P)、羧甲基纤维素钠(CMC)以7∶2∶1的比例均匀涂抹至铜箔上,60 ℃ 真空干燥12 h获得工作电极。而后将工作电极与对电极(金属锂片),隔膜(Celgard 2400),电解液共同在氩气气氛手套箱中组装成扣式电池。其中,电解液成分为1 mol 六氟磷酸锂(LiPF6)溶于碳酸乙烯酯(EC)∶碳酸二乙酯(DEC)∶碳酸甲乙酯(EMC),体积比1∶1∶1。扣式电池静置24小时后进行后续电化学实验。所有充放电测试均在新威电池测试系统(CT-3008W)上进行测试,电压窗口为0.01~3 V。
1.4 材料磁性表征
所有磁学测试均在完全无液氦综合物性测量系统(PPMS DynaCool,quantum design)上进行测试。CoO原位磁性测试电池使用与扣式电池完全一致的电极和电解液[25],在氩气手套箱中进行组装。恒流充放电和循环伏安法CV 原位磁学测试是在300 K 的温度下,0.01~3 V 的电压窗口内,以500 mA/g 的电流密度和0.5 mV/s 的扫速进行充放电循环测试,同时保持3 T磁场获取磁学信号。等温磁化曲线(Magnetic Hysteresis,M-H)测试分别在300 K及5 K的温度下,在-3 T~3 T的磁场变化区间内进行测试。零场冷/场冷(zero-field-cooled/field-cooled,ZFC/FC)变温磁化曲线测试则是在500 Oe(1 Oe=4π×10-3A/m)的恒定磁场下,5~350 K的温度范围内进行测试。
2 结果与讨论
2.1 CoO杂质相检测
首先将CoO 放入H2/Ar 中分别还原20 min 及40 min 制备不同比例还原Co 样品,记作CoO/Co@20min,CoO/Co@40min。为探测合成样品物相,对合成材料进行XRD测试。所制备的CoO、CoO@20min、CoO@40min XRD 图 谱 如 图1 所示。可见,所有物质特征峰均较好匹配面心立方(fcc结构)CoO (ICSD No.01-078-0431)。三种材料的XRD 图谱不仅无杂质峰,并且衍射峰强度相似。此结果一方面表明CoO 材料的成功制备,另一方面表明常规XRD 检测无法甄别出除CoO 以外的其他物相。
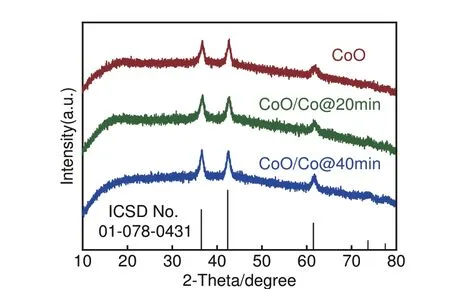
图1 CoO、CoO@20min、CoO@40min XRD图谱Fig.1 XRD patterns of CoO, CoO@20min, CoO@40min
进一步,为了明晰还原出的微量Co 对样品元素价态及晶体结构的影响,本文分别使用XPS 及HRTRM 对三种材料进行表征,结果如图2 所示。图2(a)、(b)、(c)分别为样品CoO、CoO/Co@20min、CoO/Co@40min 的XPS 图谱,三种材料的Co 2p高分辨能谱均展示出区别于Co单质与Co3+的典型的Co2+特征,能谱在786 eV 附近有一个非常明显的震激谱峰[26]。以780.2 eV及782.5 eV为中心的特征峰为Co2+的2p3/2多重分裂峰,以795.2 eV及797.5 eV为中心的特征峰为Co2+的2p1/2多重分裂峰。此外,787.7 eV及802.7 eV附近的特征峰分别为Co2+的2p3/2、2p1/2的卫星峰。三种材料X射线光电子能谱除特征峰面积有略微区别外,各个特征峰位基本一致,无法从能谱中识别出除Co2+以外的其他价态信号。同时,据图2(d)、(e)、(f) HRTEM图谱所示,三种材料晶格条纹清晰连续,晶格间距为0.246 nm 及0.15 nm 的晶格条纹分别对应CoO的(111)及(220)晶面,无其他物相的晶格条纹。综上,制备的三种材料通过XRD、XPS、HRTEM表征均仅探测到CoO 信号,即三种传统表征手段均无法有效识别出材料中还原出的微量Co单质。
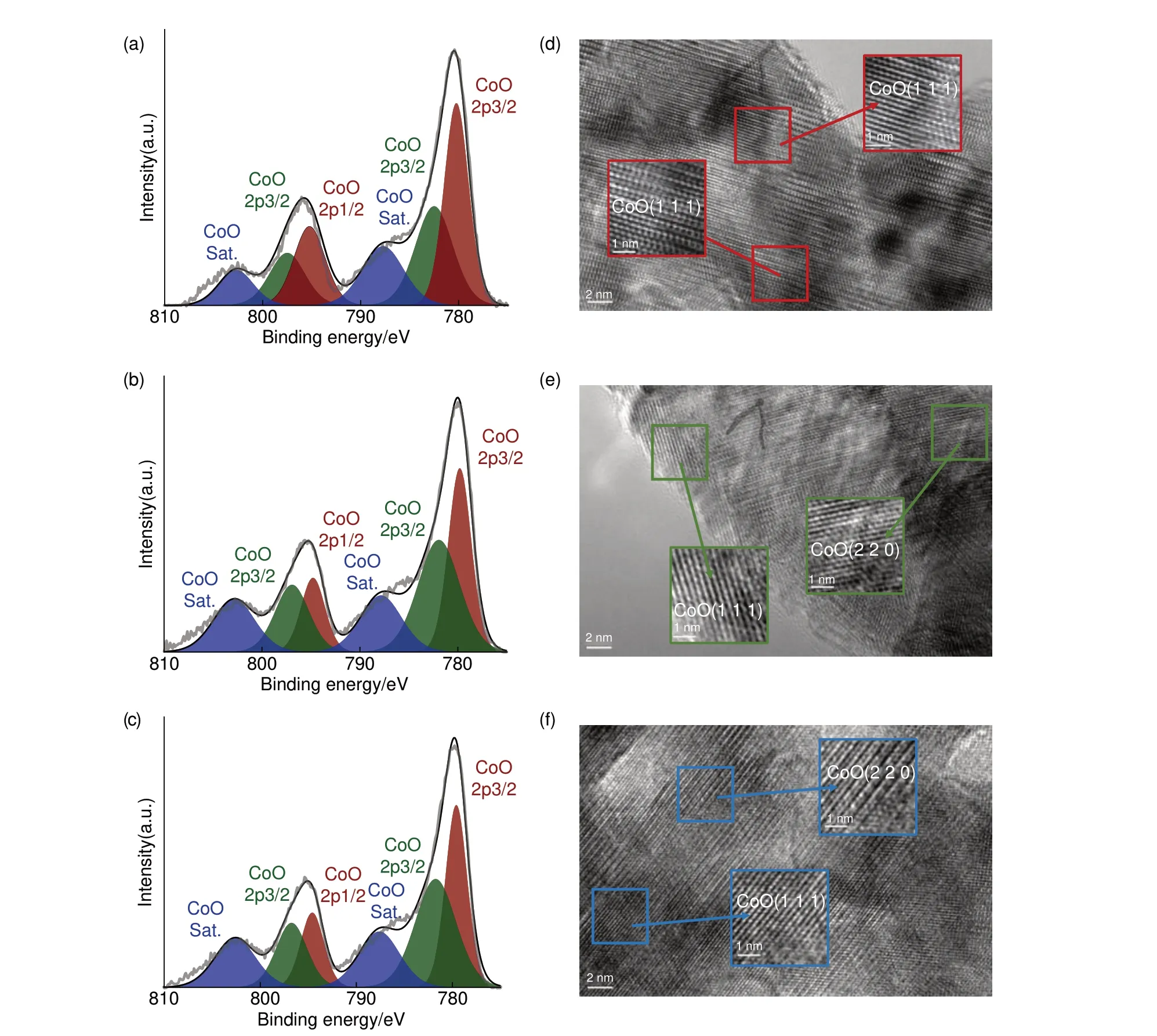
图2 所制备样品的XPS及HRTEM图谱XPS图谱 (a) CoO,(b) CoO@20min,(c) CoO@40min;HRTEM图谱 (d) CoO,(e) CoO@20min,(f) CoO@40minFig.2 XPS and HRTEM patterns of the prepared samples XPS patterns of (a) CoO, (b) CoO@20min, (c) CoO@40min; and HRTEM patterns of (d) CoO,(e) CoO@20min, (f) CoO@40min
由于传统表征手段无法检测还原出的微量Co单质,而样品磁性可反映材料物相、晶体结构、元素价态以及电子能带结构等信息,此外磁性测试对Co 这一类过渡金属极为敏感,因此本文通过磁学测试监测CoO 杂质相。等温磁化曲线(magnetic hysteresis,M-H)测试,即测量样品磁矩随外加磁场变化情况,分析测试结果——磁化曲线即可获知样品在不同磁场下的磁化行为[27]。由于不同样品的磁化曲线表现不同,所以M-H测试是一种检测与主体材料磁性不同杂质非常方便、灵敏、快捷的检测手段[28]。CoO、CoO@20min、CoO@40min 300 K下M-H 测试结果如图3(a)、(b)所示。CoO 是一种典型的反铁磁(AFM)材料,其奈尔温度受颗粒尺寸影响通常在225~293 K[29],在常温下,外界温度超过其奈尔温度(TN),晶体内能破坏材料内部磁性排列,样品由反铁磁性转变为顺磁性。纯净CoO磁化曲线如图3(a)所示,线性且无磁滞,3 T外加磁场下磁化强度仅为1.895 emu/g(1 emu=10-3A·m2)。而CoO@20 min、CoO@40min 样品磁化曲线低磁场下表现出非常明显的磁性转变,表明测试样品中存在铁磁性(FM)杂质Co的信号[30]。由于铁磁性物质本身自旋排列有序,在样品磁化后,即使外加磁场消失,仍有部分自旋保持排列,形成剩磁,则需要一个额外磁场进行消磁,该磁场称为矫顽力。CoO@20min、CoO@40min样品均有较大矫顽力,分别为674 Oe 以及874 Oe,剩磁分别为0.5155 emu/g、1.9642 emu/g。铁磁性杂质矫顽力与团簇大小相关,基于Billas 等人[31]的研究成果,可以推测随H2还原时Co 铁磁团簇逐渐变大。与此同时,随着还原时间增长,样品的磁化强度明显增大,也进一步证明了铁磁杂质含量的增多。结合CoO 磁化率及Co 饱和磁化强度,估算CoO@20min 内含0.66%的Co金属杂质,CoO@40min内含有2.27%的Co金属杂质[15]。
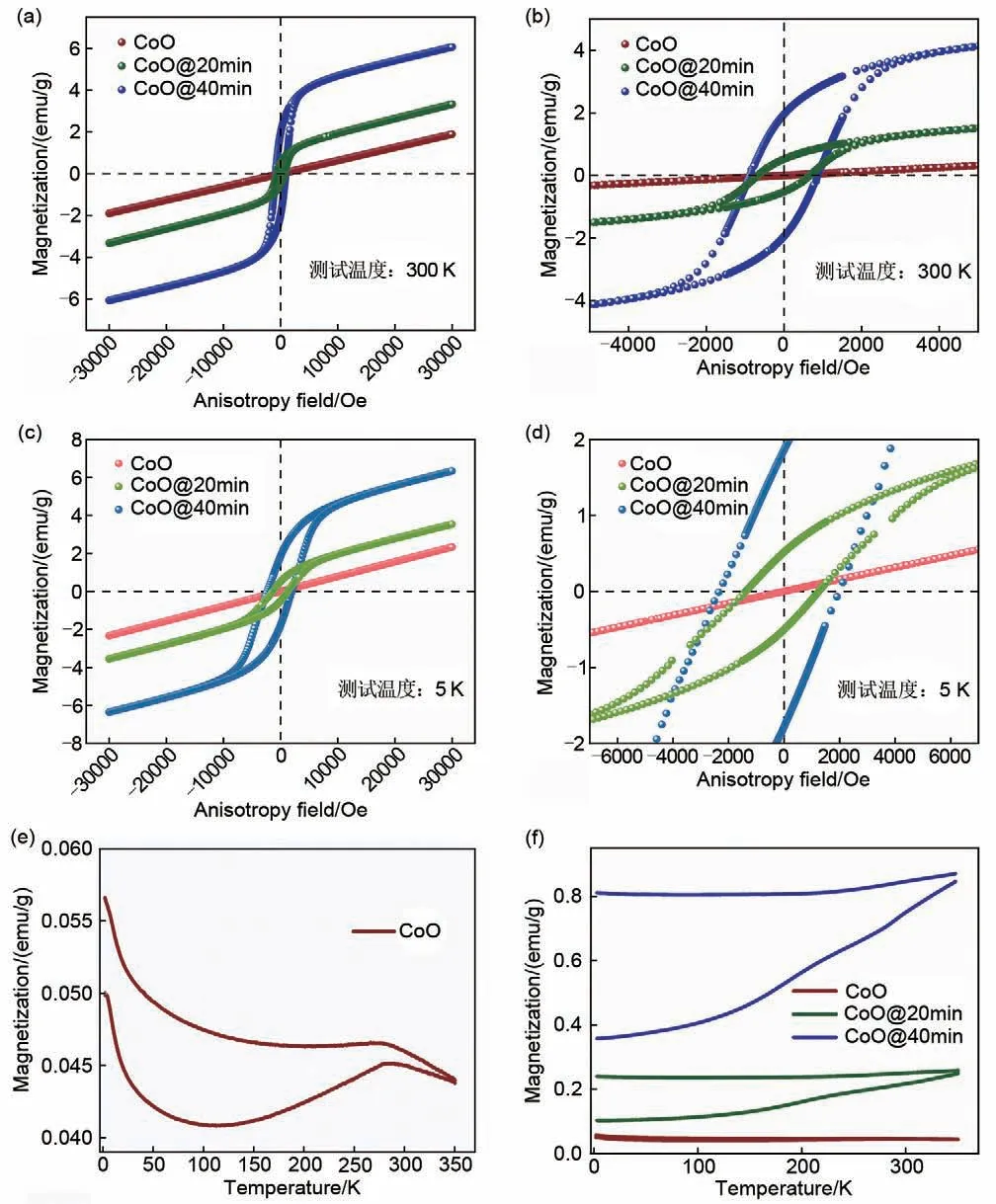
图3 样品磁性测试图谱(a) CoO、CoO@20min、CoO@40min 300 K M-H图谱;(b) 三种样品300 K M-H曲线局部放大图谱;(c) CoO、CoO@20min、CoO@40min 5 K M-H图谱;(d) 三种样品5 K M-H曲线局部放大图谱;(e) CoO 500 Oe磁场下ZFC/FC图谱;(f) CoO、CoO@20min、CoO@40min ZFC/FC对比图谱Fig.3 Samples magnetic characterization(a) CoO,CoO@20min,and CoO@40min 300K M-H curves; (b) localized magnification of 300 K M-H curves for three samples; (c) CoO, CoO@20min, CoO@40min 5 K M-H curves; (d) localized magnification of 5 K M-H curves for three samples; (e) ZFC/FC curves of CoO under 500 Oe; (f) CoO, CoO@20min,CoO@40min ZFC/FC curves comparison maps
随着温度的降低,热扰动效应明显减弱,因此可以更有效地观察到材料的基态磁性,基于此本工作进行了低温M-H 测试来观察CoO 体系内的杂质相[32]。当进行5 K M-H测试时,如图3(c)、(d)所示,反铁磁CoO磁化曲线仍然随磁场变化线性变化,且无矫顽力,在3 T外加磁场下磁化强度为2.3356 emu/g。有趣的是,CoO@20min、CoO@40min 样品在低温下出现了磁性转变,表现出了不同程度的交换偏置效应。交换偏置效应通常是指发生于铁磁(FM)/反铁磁(AFM)界面处的一种效应[33]。材料温度降至奈尔温度TN以下时,界面处反铁磁层自旋有序排列。当外加磁场变化时,铁磁层受到界面处反铁磁层的钉扎效应影响,自旋发生磁性翻转时必须额外克服这一影响,最终导致材料磁滞回线相较于零场偏移[34]。实验测得,CoO@20min 交换偏置场为60.5 Oe,CoO@40min 交换偏置场为226.5 Oe。低温M-H 测试结果证实了引入铁磁性Co 杂质,交换偏置场的增加同样验证了随H2还原时间增加,Co铁磁团簇的增加。
材料磁学性质随温度改变而改变,不同磁性材料有着不同的磁性转变温度,磁矩随温度变化趋势也不相同,所以ZFC/FC变温磁化曲线测试同样是一种非常有效、灵敏的杂质相检测手段[35]。本工作通过变温ZFC/FC测试识别杂质相与母相的特征转变温度进一步检测CoO中的杂质相。零场冷(ZFC)是指无外加磁场情况下,将样品降至低温,而后加一磁场进行升温磁化率测试;场冷却(FC)则是保持外加磁场降温,而后进行升温磁化率测试。由于ZFC 会冻结未排列自旋电子,而FC 在降温时电子已经在外场作用下完成排列,所以二者曲线会有明显差异。比较ZFC/FC 曲线可以获知磁性相变温度、自旋玻璃态及超导临界温度等信息[36]。CoO样品ZFC/FC 曲线由图3(e)所示,TN=283 K,ZFC/FC曲线呈现明显的尖峰,对应CoO反铁磁奈尔温度。在整个温度范围内,ZFC/FC 曲线不重合显示体系内可能存在铁磁团簇导致的自旋玻璃态[37]。而CoO@20min、CoO@40min 由于存在微量Co 纳米颗粒,磁化率相较于CoO 样品明显增强近一个数量级,ZFC/FC 曲线在整个测量温区内没有观察到明显的反铁磁相变,FC 曲线反而呈现出类似铁磁性饱和特征[38]。
为了探究微量Co杂质对CoO材料储能性能的影响,本文作者进行了相应的电化学实验。图4分别展示了CoO、CoO@20min及CoO@40min的循环伏安测试CV 图谱,电压窗口为0.01~3 V,扫速为0.5 mV/s。三种材料CV 曲线形状相似,峰位一致,但仍可以观察到两个显著差异:①CoO@20min 和CoO@40min 样品CV 曲线相较于CoO 材料从循环第二圈开始,0.65~1.5 V 之间的还原峰交叠明显减少,说明微量Co 的引入减小了材料的极化;②CoO@20min和CoO@40min在2.15 V的氧化峰面积明显高于CoO,说明引入Co 后材料氧化还原更加充分。图5(a)首圈恒流充放电图谱展示了微量Co 对材料首圈库仑效率的影响。CoO 首圈库仑效率为74.3%,CoO@20min 为80.99%,CoO@40min则为83.77%,这说明Co的引入显著提升了CoO 首圈库仑效率,这种变化可能归因于Co单质促进了SEI膜的可逆形成分解。在微量还原后,CoO材料倍率性能也有显著提升,根据图5(b)倍率电流测试图谱所示,CoO 样品在1 A/g 电流密度下容量即出现明显衰减,而CoO@20min样品则在2 A/g 电流密度下容量才开始出现微弱衰减,CoO@40min样品倍率性能最优,并且全程保持容量逆衰减趋势,且不同电流密度间容量差异最小。如图5(c)所示,大电流长循环测试中,CoO@40min样品表现出最优的循环稳定性,初始容量最大为1021.54 mAh/g,且出现容量逆衰减,循环50圈后容量保持率为116.59%;CoO@20min样品表现次之,循环50 圈后容量保持率为114.17%;未还原样品CoO表现最差,初始容量为908.4 mAh/g,循环50圈后,容量衰减至518.9 mAh/g,容量保持率仅为57.12%。综上所述,微量Co 的引入对CoO储能材料极化、首圈库仑效率、倍率性能及大电流密度下的循环稳定性均有较大影响。因此,甄别检测出主体材料中的微量杂质相十分重要。
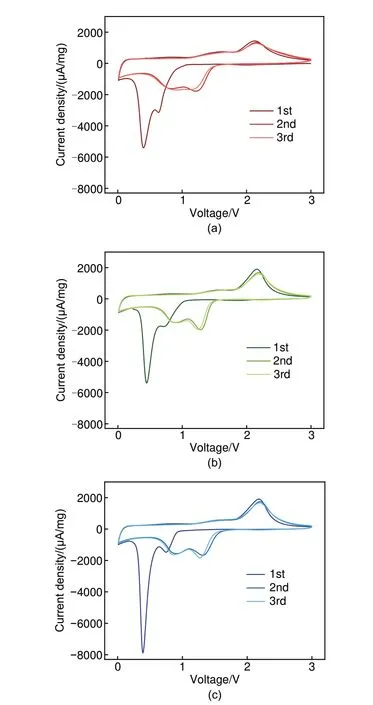
图4 (a) CoO、(b) CoO@20min、(c) CoO@40min 样品0.5 mV/s扫速下循环伏安测试CV图谱Fig.4 Cyclic voltammogram tests of (a) CoO,(b) CoO@20min, and (c) CoO@40min at a scan rate of 0.5 mV/s
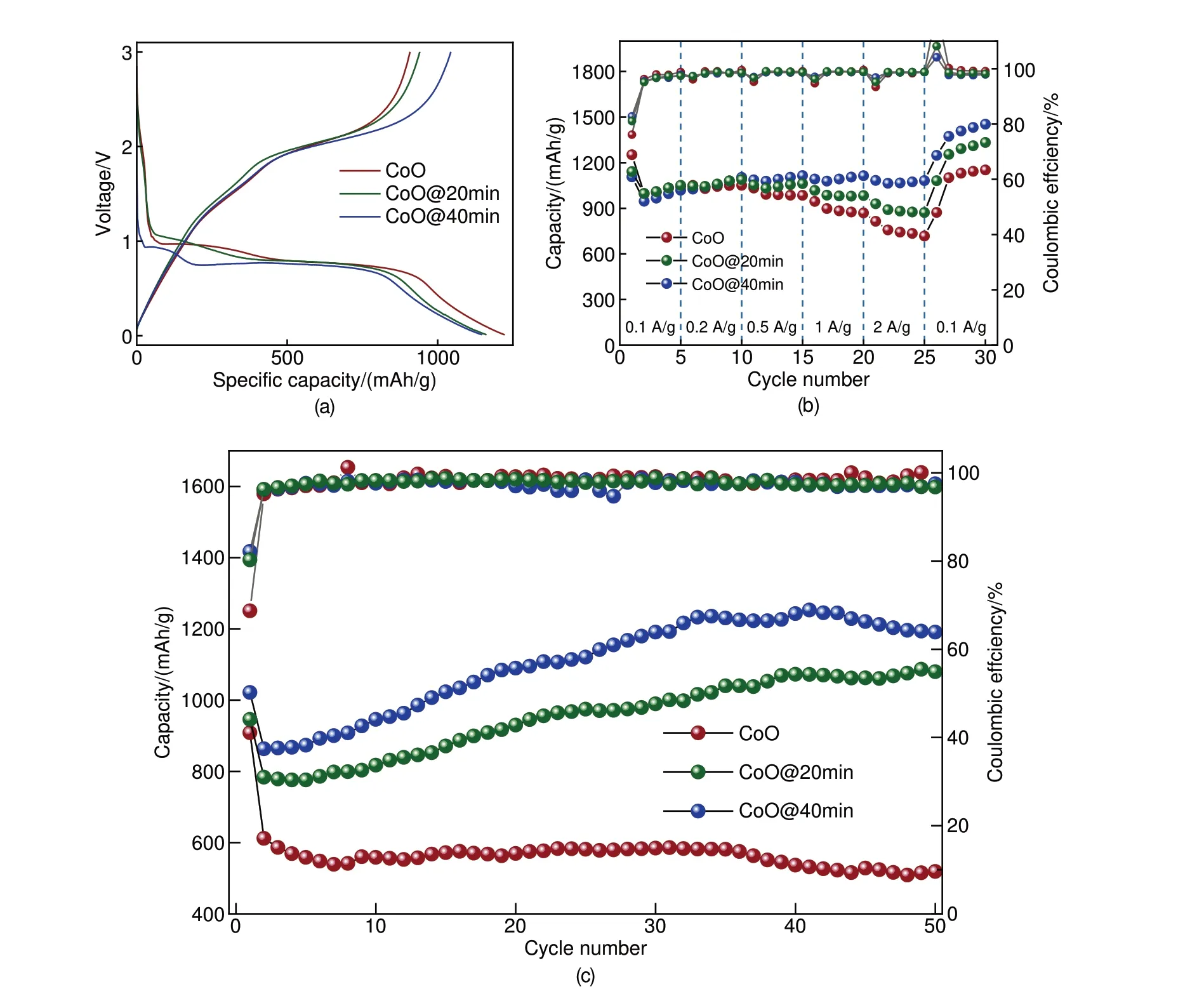
图5 CoO、CoO@20min、CoO@40min 电化学测试图谱(a) 首圈恒流充放电测试图谱;(b) 倍率电流测试图谱:(c) 2 A/g 电流密度下长循环充放电测试图谱Fig.5 Electrochemical characterization of CoO, CoO@20min, CoO@40min(a) galvanostatic Charge-Discharge (GCD) profiles for the first cycles; (b) rate capabilities at different current densities; (c) l.ong-term cycle performance at a current density of 2 A/g
2.2 CoO界面储能探测
为探测CoO 低电压区间的储能机理,本工作首先选取不同低电压电位进行非原位极片XRD 检测,以识别低电压区间物相变化以及结构相变,结果如图6(a)所示。经过与图6(b)非原位空铜箔XRD对比,可见选取的5 个电位的XRD 均仅有铜信号(ICSD No.00-003-1005),无其余活性物质衍射峰峰位。为确定具体原因,对充分锂化的CoO (0.01 V)进行非原位M-H测试。磁化曲线为典型超顺磁材料磁滞回线,此状态下的Co 单质颗粒尺寸小于临界尺寸,呈超顺磁性,可用郎之万方程拟合Co 平均颗粒尺寸[39],结果如图6(c)所示。经拟合,Co平均颗粒尺寸为1.792 nm,此尺寸已超出传统XRD 检测极限:2~2.5 nm,所以非原位XRD无除铜以外任何活性物质峰位[40]。综上,传统XRD检测无法表征CoO低电压区间物相改变。
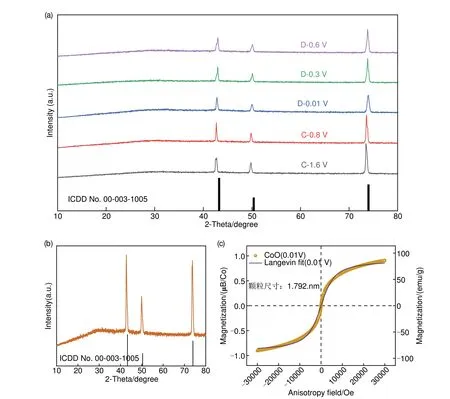
图6 (a) CoO低电压区间非原位XRD测试图谱;(b) 空白铜箔XRD图谱;(c) CoO锂化后 (0.01 V)磁滞回线(黄色)及相应郎之万拟合曲线(紫色)Fig.6 CoO (a) ex-situ XRD patterns in the low voltage interval of CoO; (b) XRD pattern of blank copper foil; (c) Magnetic hysteresis curves of the CoO electrode after (yellow) the lithiation process (0.01 V), and the corresponding Langzhiwan fitting curve (purple)
原位磁学测试实时、无伤检测充放电过程中的磁化强度变化,比较电化学曲线以及磁性曲线,可以获知电化学过程中的物质转化、结构改变、电子转移等信息,从而分析电化学储能机理,发掘全新的储能机制[13]。特别的,原位磁学测试可以检测电极材料界面处的电荷转移情况,从而分析能源科学中的界面储能问题。CoO全区间原位磁学CV测试如图7所示,可见首圈从开路电压放电至0.17 V 有一明显的磁性上升过程,磁化强度达130.9 emu/g,此过程对应于CoO 还原为Co。而后磁化强度略微下降再上升,直至充电到1.75 V 时磁性急速下降,这源于Co 氧化为CoO。在第二圈循环后,还原电位移至1.58 V。为更细致检测低电压区间储能机理,本文作者针对第二圈电化学循环过程进行原位磁学恒流充放电测试。
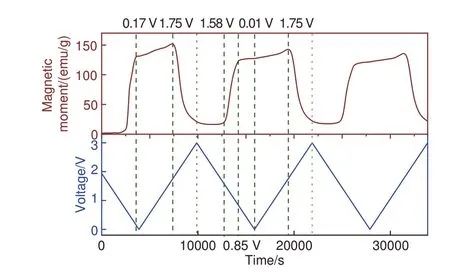
图7 CoO 原位磁学CV测试图谱,外加磁场为3 T,扫速为0.5 mV/sFig.7 In situ magnetometry CV test pattern of CoO under an applied magnetic field of 3 T and at a scan rate of 0.5 mV/s
CoO第二圈原位磁学恒流充放电测试结果如图8所示,磁性放电至1.6 V 后急速上升,考虑到极化等因素影响,该结果与原位磁学CV 测试匹配。而后材料放电至1.1 V 后,磁性上升趋势变缓,直至0.625 V 到达区域磁性最低点。这种磁性变化趋势可以用界面处空间电荷储能机制解释[19],空间电荷机理如图9(a)所示。根据Maier 的空间电荷模型,当CoO还原为Co金属后,部分电子可储存于电子导体Co表面,而Li+则可储存于Li2O等离子导体表面,贡献额外容量[41-42]。电子注入前,Co d轨道能带中自旋朝上电子多于自旋朝下电子,而由于Co纳米颗粒费米能级处自旋朝下态密度更高,因此后续注入电子自旋极化朝下更多,与还原引起的磁性上升竞争导致放电后期磁化强度变缓及下降[43-44]。
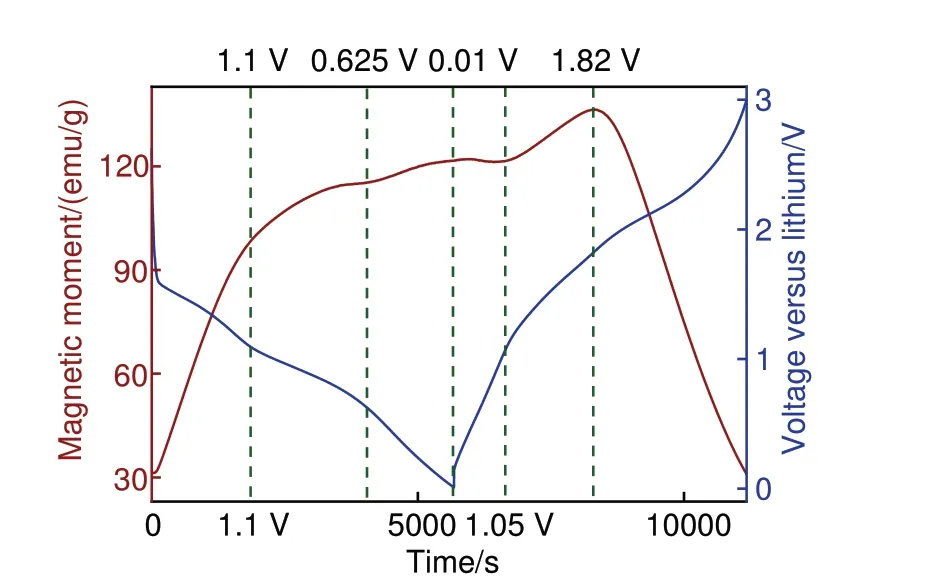
图8 CoO 第二圈原位磁学恒流充放电测试图谱,外加磁场为3 T,电流密度为500 mA/gFig.8 In situ magnetometry GCD test pattern of CoO for the second cycles under an applied magnetic field of 3 T and at a current density of 500 mA/g
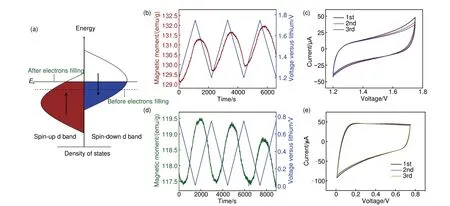
图9 Co/Li2O界面自旋极化机理及分区原位磁学CV测试示意图(a) Co颗粒表面自旋极化电子填充前后态密度变化示意图,EF为费米能级;(b) 1.2~1.7 V,(d) 0.01~0.75 V电压窗口内,CoO原位磁学CV测试图谱;(c)、(e) 为对应电压窗口内的循环伏安测试CV图谱;外加磁场为3 T,扫速为0.5 mV/sFig.9 Schematic of spin-polarization mechanism and zoned in situ magnetometry CV test at Co/Li2O interface(a) schematic of the density of states (DOS) changes before and after spin-polarized electrons filling on the Co particle surface, and the EF is the Fermi energy; In situ magnetometry CV test pattern of CoO over the potential range of (b) 1.2—1.7 V , (d) 0.01—0.75 V respectively; (c)、(e) their corresponding CV patterns; The applied magnetic field is 3 T and the scan rate is 0.5 mV/s
后续可见磁性异常重复上升直至0.01 V,Tarascon教授等[45-46]对CoO锂离子电池充放电过程进行了系统的TEM 探究,他们提出:低电压时,可能发生了电子转移到碳酸烷基酯分子上形成自由基负离子(该自由基负离子由Co表面吸附的锂离子稳定)这样一个SEI膜的形成过程,并且在高电压时SEI膜分解。这样的物理图像与本工作监测到的原位磁学信号十分相符,所以本文作者推测此阶段异常磁性上升信号可能源于活性物质表面SEI膜的形成利用了界面处的自旋极化电子[47]。而后充电至1.05 V,磁性下降,可归因于SEI膜的分解。再充电至1.82 V,自旋极化电子被释放,磁性上升,后续充电至3 V,Co氧化为CoO,磁性下降。
CoO还原构筑储存空间电荷界面,SEI膜生成提取自旋极化电子,原位磁学测试通过磁学信号真实、精准地表征CoO 低电位区间界面储能过程,反映出Co 氧化还原、空间电荷、SEI 膜三者相互竞争、相互成就的关系。此外,CoO 理论容量为715 mAh/g[48],而实际恒流充放电测试发现放电容量可达995.29 mAh/g(第二圈),远高于其理论容量,并且后续伴随明显的容量逆衰减行为,原位磁学对低电压区间界面储能的探测揭示了这一现象的根源,即低电压区间的空间电荷嵌入脱出及SEI膜形成分解共同贡献了额外容量。
为确保结论翔实可靠,本文还针对上述分析的不同电压分区进行了原位磁学CV 测试。分别选用充电过程中1.2~1.75 V,0.01~0.75 V 进行测试,结果如图9(b)、(c)、(d)、(e)所示。首先,如图9(c)、(e)所示,各分区CV曲线呈矩形,表明空间电荷及SEI 膜储能均为电容行为。此外,如图9(b)所示,1.2~1.75 V 区间内避开了Co 氧化还原区间及SEI膜区间,磁性随电压上升而上升,随电压下降而下降,这与自旋极化空间电荷趋势完全一致。而在0.01~0.75 V电压范围内,如图9(d)所示,磁性变化趋势与空间电荷磁性变化趋势相反,证实SEI膜的形成分解利用释放了自旋极化电子。基于分区原位磁学CV 测试结果,可见空间电荷及SEI 膜两种储能机制在循环过程中高度可逆。
3 结 论
本工作首先通过高低温M-H,ZFC/FC 磁学测试定量、精准、快速地探测出CoO中的微量金属Co单质杂质相,其中,CoO/Co@20min内含有0.66%的Co单质杂质,CoO/Co@40min内含有2.27%的Co 单质杂质。经后续电化学性能表征发现,正是这些无法被XRD、XPS、HRTEM等传统表征手段探测出的微量Co 却能显著减少CoO 主体材料的极化,提升首圈库仑效率(74.3%~83.77%),极大提高了CoO 材料的大电流下的循环稳定性(2 A/g 50 圈容量保持率116.59%)。此外,本工作还结合原位磁学CV 测试、恒流充放电测试,直观表征CoO 低电位区间复杂的界面储能过程,揭示出Co氧化还原、空间电荷嵌入脱出、SEI形成分解三者之间相互竞争、相互成就的关系,解释了CoO 远超其理论容量限制的额外容量现象,超越了非原位XRD 物相检测的测试极限。本工作成功展示了磁学测试在识别微量杂质相及监测复杂界面储能中的应用潜力,可广泛应用于各种过渡族金属化合物电极材料,不仅加强了材料科学与磁性研究的交叉,还为新型电化学储能技术的开发提供了新的思路。
