融合多组学数据的乳腺癌生存期预测模型研究
2024-01-26方秋莲周仪璇
方秋莲,周仪璇,伍 幸
(中南大学 数学与统计学院,湖南 长沙 410083)
0 引言
乳腺癌是一种发生于女性身上的恶性肿瘤[1],是目前女性中发病率最高的恶性肿瘤[2].研究表明,大多数已确诊乳腺癌的患者可以通过及时有效的治疗被治愈或者延长生存期[3].当前乳腺癌诊疗研究的发展趋势是根据患者的遗传和临床信息对其进行精准预后,并基于预测结果为其制定个性化的医疗方案.生存期预测是乳腺癌预后的重要研究内容之一[4].乳腺癌的发生和发展是一个复杂、多阶段和连续的过程,不仅受遗传因素(基因突变、表观遗传变化、细胞生物环境、个体特有特征)影响,也受患者生活环境因素影响,涉及多种生物分子水平的变化[5].近年来,得益于大规模组学数据的收集和计算方法的发展,从多个生物学层面解读乳腺癌疾病机制的研究取得了重大进展.由高通量实验技术产生的海量数据统称为组学数据,组学数据为细胞的分子机制提供了详细描述,不同层次组学数据在全基因组的分布状态与乳腺癌的发生、发展、预后之间均存在关联关系[6].目前的研究趋势是综合运用多个组学的信息对乳腺癌患者进行预后.与单一组学数据相比,多组学数据可以提供更全面的基因信息,利于更好地解释分类结果[7]、改善预测效果[8]、理解复杂的分子机制[9].然而,多组学数据的融合面临许多阻碍.一方面,组学数据自身具有缺失、类不平衡、噪声大、复杂及维数灾难等特点,这使计算变得困难[10].另一方面,不同来源的数据集具有异质性,具体表现为数据分布、类型、规模的差异.组学数据的这些特点为数据的融合过程带来了挑战.
现有的数据融合方法可按融合的时间点分为5类:早期、混合、中期、晚期和分层融合[11].早期融合即在建模前将所有数据集拼接为一个大型矩阵,这种方法思想简单且易解释,但会得到一个更加复杂高维的矩阵,增加了后续建模难度[11].混合融合是先将原始数据转换为低维、少噪声的简单表达,然后融合数据的简单表达[11-12].这类方法在进行融合前不仅降低了数据的维度,还减弱了数据集间的异质性,如数据规模.中期融合即直接融合未经转换或拼接的数据集并输出可用于后续分析的新的数据表达,这类融合方法假定所有数据集可投影到一个公共的隐空间[11,13].晚期融合也可称为模型融合,即先分别对所有数据集建模,然后组合模型预测结果[11,14].分层融合即在神经网络的连接结构中引入分子通路关系的先验信息,可以在模型中体现分子层面的模块结构[11,15].目前尽管已经有一些利用多组学数据预测乳腺癌患者生存期的研究,但是使用基于模型的晚期融合方法来提升预测精度的研究较少.本文采用基于模型的晚期融合方法,其优势在于可以根据每一个数据集的特点分别选择最合适的模型,充分考虑了数据集的异质性,且无须直接整合异质的数据.在融合环节,本文采用加权平均法组合模型预测结果,其中权重的取值由模型效果和预测结果间的互信息决定,这样的组合方式可以改善预测效果,增强预测结果的可解释性.
1 数据介绍及预处理
1.1 组学数据介绍
本文使用的数据来自 METABRIC数据库[16],包括1 981位乳腺癌患者的临床信息数据、1 904位乳腺癌患者的基因表达信息数据以及2 173位乳腺癌患者的拷贝数变异信息数据,这3个数据集可通过共同字段——“PATIENT_ID”和“Hugo_Symbol”(均代表患者编号)进行连接,患者的生存时间变量包含在临床信息数据集中.定义患者的生存时间是否大于5年为因变量,剔除随访期小于5年且生存状态为存活的样本,最终得到的基因表达数据集样本量为1 844,临床信息数据集和拷贝数变异数据集样本量均为1 917,所有患者的最短生存期为0.1个月,最长生存期为29.60年,中位生存期为9.96年.
组学数据具有缺失、高维等特征,因此,需要先进行数据清洗、缺失值插补、特征选择等数据预处理工作.
1.2 缺失值处理
本文首先剔除数据集中含缺失的样本得到无缺失的数据集,然后在无缺失的数据集上人工随机生成缺失,缺失的生成比例即原始数据集中该特征的缺失比例,然后基于此数据集从多个缺失值插补方法中选出效果最好的方法,并将该方法用于原始数据的缺失值插补.本文用到的缺失值插补方法有:随机森林预测插补法、K最近邻(KNN)预测插补法、简单均值众数插补法以及基于K-means聚类的均值众数插补法.对于数值型特征,本文采用实际值与插补值的均方误差来衡量插补效果,对于类别型特征,采用实际值与插补值的F1值来衡量插补效果.
1.3 特征选择
本文的特征选择过程均是在单个数据集上进行,分为3步:
1)采用假设检验法分别对3个数据集的特征进行初筛,选择在长生存期和短生存期两类样本上的分布具有显著差异的特征.对于数值型特征:首先分别对两类样本上的特征进行Shapiro正态性检验和Levene方差齐性检验.若两类样本上的特征取值均服从正态分布且具有等方差性,则对特征进行方差分析.若两类样本上的特征取值不全服从正态分布或不具有等方差性,则对两类样本上的特征进行Wilcoxon秩和检验,选择方差分析或Wilcoxon秩和检验显著的特征,将其纳入特征子集[17].对于类别型特征:首先构建特征与因变量之间的列联表,根据列联表单元格中的计数来进行相关检验:若全部单元格计数T≥5且样本总数n≥40,则用Pearson卡方检验;若存在单元格计数1≤T<5且样本总数n≥40,则用连续性校正的卡方检验;若存在单元格计数T<1或n<40,则用Fisher精确检验[17].
2)对第一步选出的特征建立随机森林模型,计算每个特征的基尼重要性和基于排列的重要性,剔除重要性为0的特征,综合两种特征重要性对剩下的特征进行排序.
3)将上述特征依序逐个加入Logistic回归模型,若加入的特征能使模型的AUC提升10-6,则将加入的特征保留下来,否则将其剔除.
1.4 数据标准化
完成缺失值插补及特征选择后,在训练模型之前,需要对数据进行标准化.这是因为部分机器学习模型如支持向量机、KNN等是基于距离度量进行样本分类和预测的,而距离对量纲和取值范围非常敏感.数据标准化的过程如式(1)所示:
(1)
式中:X为原始特征;Xmean为X的平均值;Xstd为X的标准差;z为标准化后的特征.
2 正类预测概率加权融合模型构建
对数据进行预处理后,为了充分利用每一个数据集的信息,本文先为单一数据集选取最优分类模型,模型的融合是通过对3个数据集上的最优模型的正类预测概率进行加权求和实现.图1展示的是本文构建多组学数据融合模型的流程.
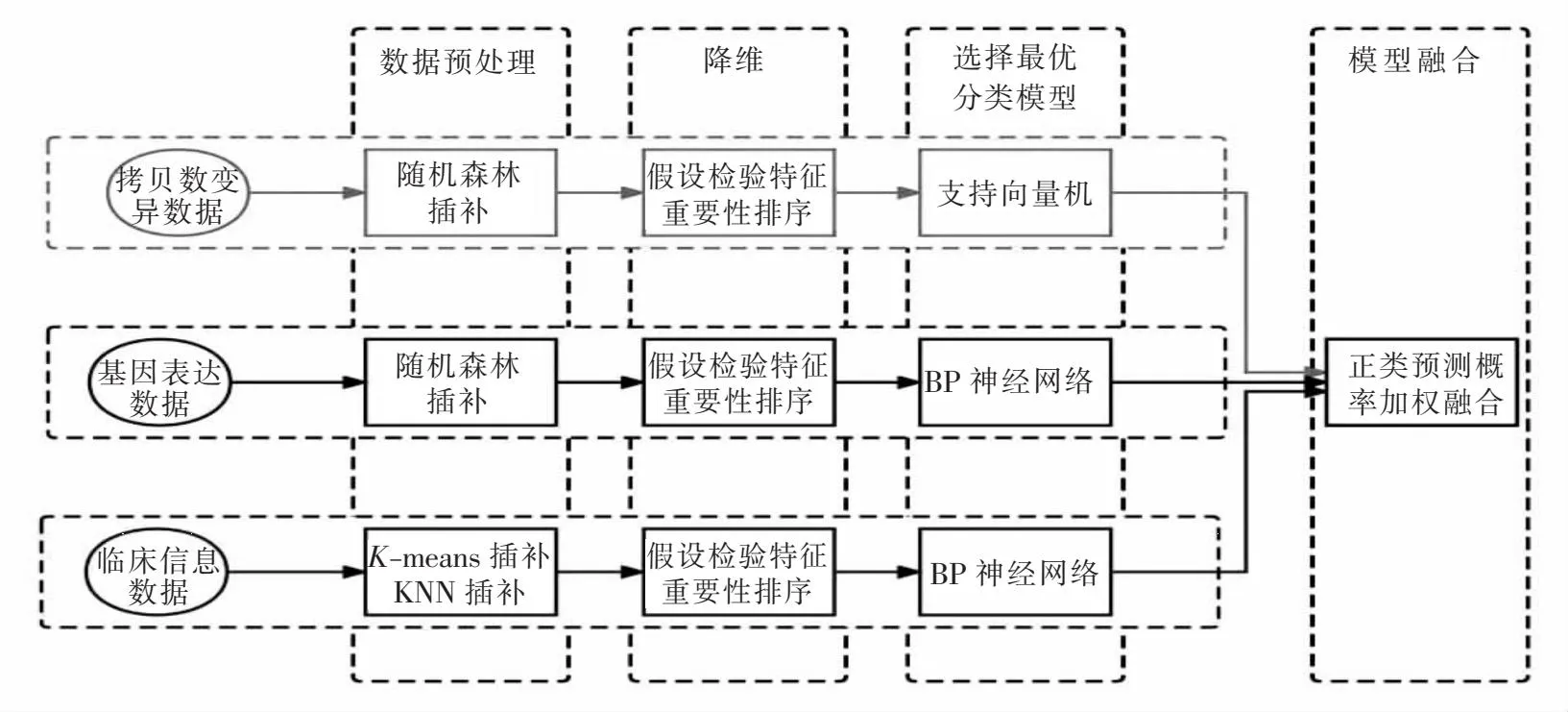
图1 融合模型构建流程Fig.1 Process of integrating model building
2.1 最优分类模型选择
本文采用Logistic[18]、支持向量机[19]、随机森林[20]、Xgboost[21],以及BP神经网络[22]5种机器学习分类算法分别对基因表达、拷贝数变异和临床信息数据集建立5年生存期预测模型,并以模型在训练集上的AUC值为评价指标,分别为每种数据集选择一个最优预测模型.
2.2 数据融合策略
在确定了3种数据集上的最优预测模型后,本文通过加权最优模型的预测结果实现数据融合,并在融合时给效果更好且包含更多信息的模型赋予更高的权重.
假设ge_prob,cli_prob,cnv_prob分别表示基因表达、临床信息和拷贝数变异数据集上最优模型的正类预测概率,即模型预测的样本生存期大于5年的概率,定义融合模型的预测结果final_weight_prob为:
final_weight_prob=a·ge_prob+b·cli_prob+c·cnv_prob.
(2)
若final_weight_prob大于0.5,则表示预测生存期大于5年,若final_weight_prob小于0.5,则表示预测生存期小于5年.a,b,c为权重参数,当a=b=c=1/3时,融合结果为等权重的正类预测概率加权平均.然而,由于3个模型的效果及其所利用信息存在差异,其对最终正确预测的贡献是不同的,故其对应的权重也应不同.
设G、C、V是基因表达、临床信息、拷贝数变异数据训练集上的预测结果,其取值g、c、v为0或1.令I(G,V)、I(G,C)、I(C,V)表示G、C、V间的互信息,其计算公式分别如式(3)~式(5)所示,由互信息的定义——两个随机变量共享的信息量,可知两个预测结果的互信息值体现了其对应模型共享信息的多少,互信息值越高,说明这两个模型共享的信息量越大,即其中任一模型所含的信息越少.
(3)
(4)
(5)
令AUCge、AUCcli和AUCcnv表示模型在训练集上的AUC值,定义A、B、C的计算公式如式(6)~式(8)所示:
(6)
(7)
(8)
权重参数a,b,c的计算公式分别如式(9)~式(11)所示,可见效果越好且跟其他两种模型共享信息越少的模型,其预测概率将具有越高的权重.
(9)
(10)
(11)
3 基于正类预测概率加权数据融合的乳腺癌生存期预测
3.1 数据预处理
首先,采用1.2节所述的5种缺失值插补方法对3个数据集中的缺失值进行插补,选定的插补方法如表1所示.

表1 选定的缺失值插补方法
然后分3步进行特征选择,剔除了方差小于0.1的特征后,采用假设检验、特征重要性排序和剔除冗余特征,得到3个数据集的最优特征数目如表2所示.
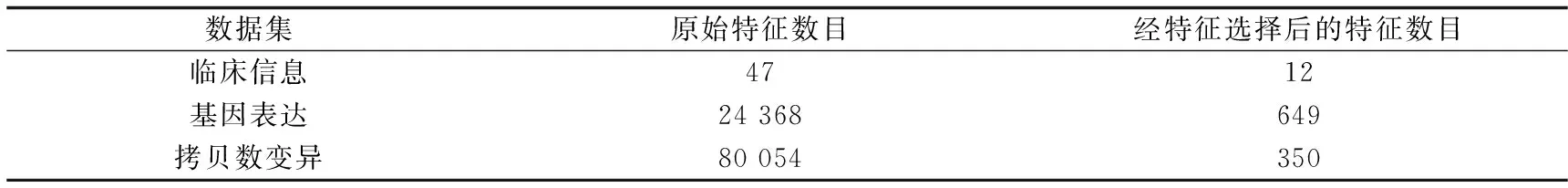
表2 3个数据集的最优特征数目
3.2 模型评价指标与调参策略
本文采用基于混淆矩阵的敏感度、特异性、准确率、ROC曲线和AUC值来评价模型分类效果.敏感度、特异性、准确率的计算公式分别如式(12)~式(14)所示.
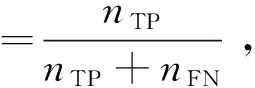
(12)
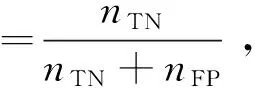
(13)
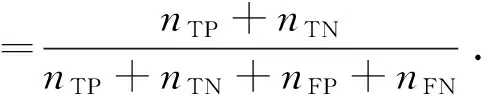
(14)
式中,nTP、nTN、nFP、nFN分别为真正例、真负例、假正例、假负例个数.
本文采用分层抽样的方式随机抽取80%的样本作为训练集,20%的样本作为测试集.在训练集上采用分层五折交叉验证、随机网格搜索以及网格搜索的方式调整模型的参数.
3.3 最优模型选择
本文先分别对所有数据按照式(1)进行标准化,然后在基因表达、临床信息和拷贝数变异数据集上训练分类模型.图2中的3幅图分别为5种分类模型在3种数据对应训练集上的ROC曲线.在基因表达数据集上,BP神经网络的AUC值最高,为0.826 8;在临床信息数据集上,BP神经网络的AUC值最高,为0.757 7;在拷贝数变异数据集上,支持向量机的AUC值最高,为0.686 0.以AUC值为主要评价指标,确定基因表达和临床信息数据集的最优分类模型为BP神经网络模型,拷贝数变异数据集的最优分类模型为支持向量机模型.
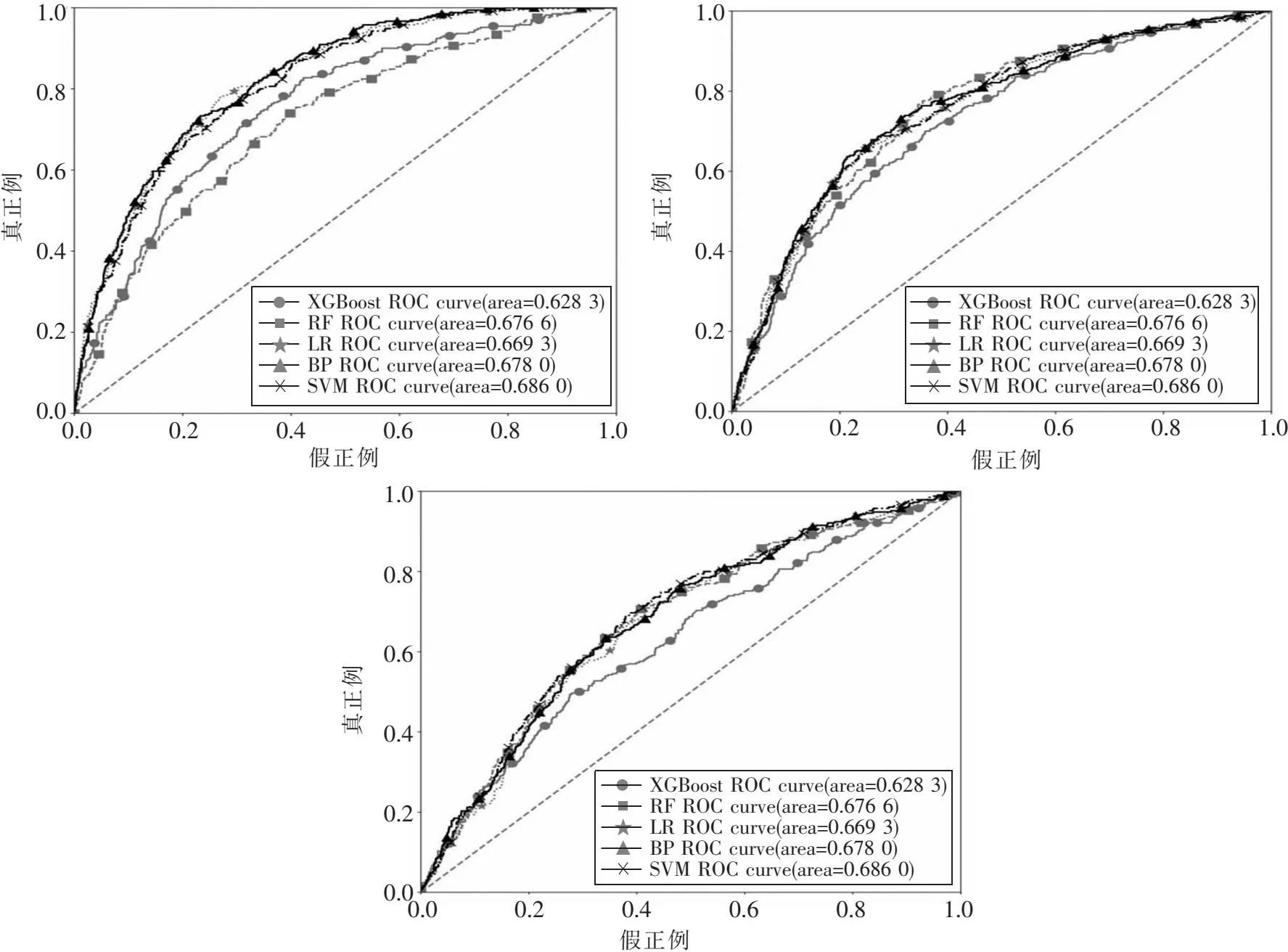
图2 5种分类算法在3个训练集上的ROC曲线及AUC值:(a)基因表达数据;(b)临床信息数据;(c)拷贝数变异数据 Fig.2 ROC and AUC of 5 types of classifiers for the three train sets:(a)Gene expression dataset;(b)Clinical dataset;(c)Copy number variation dataset
3.4 融合模型应用及其性能比较
为验证正类预测概率加权融合模型的有效性,本文综合对比基于单一数据集的模型和融合模型在测试集上各评价指标的结果.表3展示了各模型在测试集上的敏感度、特异性、准确率和AUC值.其中BP_gene和BP_cli分别表示基于基因表达和临床信息数据集预测的BP神经网络模型,SVM_cnv表示基于拷贝数变异数据集预测的支持向量机模型,equal_fusion表示等权重的正类预测概率加权融合模型,weight_fusion表示正类预测概率加权融合模型,其中权重参数a=0.275 0、b=0.488 1、c=0.236 9.敏感度最高的是基于拷贝数变异数据集预测的支持向量机模型,其敏感度为0.536 6;特异性最高的是正类预测概率加权融合模型,其特异性为0.968 6;准确率最高的是等权重的正类预测概率加权融合模型,其准确率均为0.813 0;AUC值最高的是正类预测概率加权融合模型,其AUC值为0.815 6.与基于单一组学数据集的模型相比,基于多个数据集的模型具有更高的特异性、准确率和AUC,说明融合多组学数据可提升预测效果;与等权重的正类预测概率加权融合模型相比,加权融合模型的特异性和AUC更高,说明本文提出的权重是有效的.
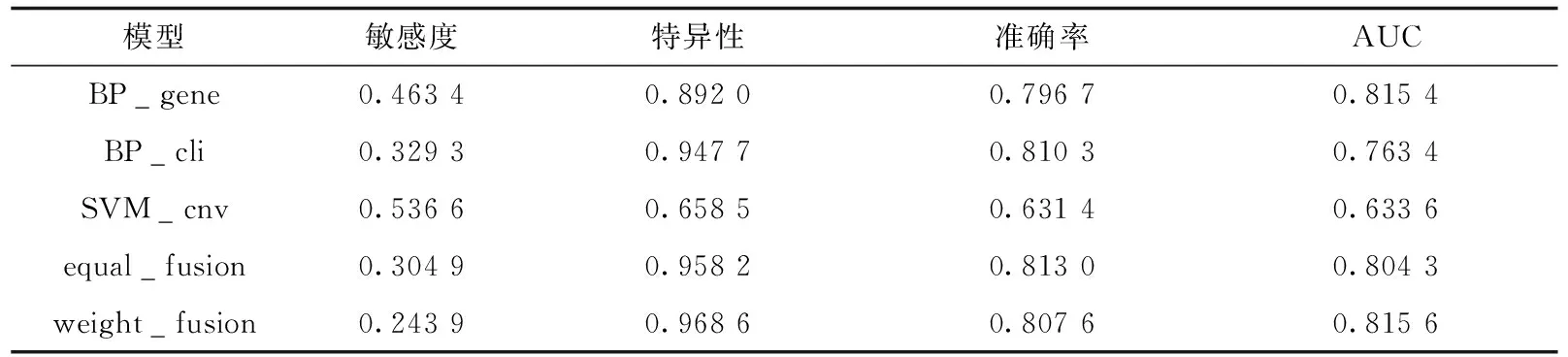
表3 各模型在测试集上的评价指标结果
4 结论
通过高通量技术获得的大量组学数据为个性化诊疗奠定了基础.本文选取基因表达、临床信息和拷贝数变异数据进行分析,分别对3个数据集进行数据清洗、缺失值插补、特征选择、最优分类模型选取,并通过求3个最优模型的正类预测概率的加权平均得到融合模型预测结果.正类预测概率加权融合模型不仅同时利用了3个数据集的信息,还在确定权重的过程中综合考虑了各个模型的准确度和它们之间的互信息.本文通过实例证明了正类预测概率加权融合模型具有良好的分类效果,其预测结果可为医生制定高效、低副作用的治疗方案提供参考.正类预测概率加权融合模型仍有提升空间,例如考虑非随机缺失,应用更先进的降维、分类算法,融合更多种类的组学数据及提升预测效果等.
