微波辐射下La-Mn-Co金属氧化物的甲苯降解性能
2024-01-26游志敏蒋思成程燊昱
游志敏,蒋思成,程燊昱,唐 凝
(湘潭大学 环境与资源学院,湖南 湘潭 411105)
0 引言
挥发性有机化合物(VOCs)是一类室温条件下沸点低于260 ℃的有机污染物[1],大多数VOCs具有易扩散、易挥发、毒性强等特点,被认为是大气污染的主要因素之一[2-3].甲苯由于存在苯环,在正常环境中结构稳定,室温条件下容易挥发,常常通过呼吸作用被人体吸收,对人体产生毒性[4].因此,甲苯通常作为VOCs中具有代表性的污染物用于研究.
目前VOCs末端治理大致可分为非破坏性技术和破坏性技术[5],在非破坏性去除途径中包括吸附、吸收、冷凝和膜分离等技术[6];破坏性技术则又可分为生物法、光催化降解法、等离子体降解法和催化氧化法等.而微波热催化作为催化氧化法的一类,与常规的热传导加热方式不同,由吸波介质吸收微波后通过内部损耗作用将微波辐射能转化为热能,属于选择性加热方式,具有加热快速高效、无二次污染、节能、清洁、操作简单等优点[7].此外,由于微波加热下的热点效应,会导致催化剂内部吸波活性位点的实际温度远远高于周围环境温度,这在催化氧化降解污染物的过程中具有很大的优势.Ding等[8]采用共沉淀法成功合成了一系列具有尖晶石结构的铜锰氧化物,分别测试了催化剂在微波场以及常规热场的活性差异,结果表明当铜锰比为1,煅烧温度为400 ℃时制备的Cu1Mn1-400催化剂在微波场中具有最高的甲苯降解活性,其表观活化能低于常规加热下的活化能,在155 ℃温度条件下便可以实现100%的甲苯转化率,极大地降低了常规热场中甲苯完全转化所需要的温度(>200 ℃).因此,以微波热场替代传统热源能很好地降低反应所需要的能量,加快催化反应进程,拥有很好的应用潜力.
微波催化氧化的关键与难点在于材料的选择,既要满足良好的催化活性,又要保证材料具有优异的微波吸收性能.与贵金属材料相比,单一的金属氧化物对VOCs的催化降解活性普遍偏低,若通过一定的制备方法将多种金属氧化物进行复合,使其中的两种及以上金属元素产生协同作用,可加快气相氧的循环.此外,不同金属元素复合使催化剂表面产生更多的缺陷,从而增加反应的活性位点[9].已有研究表明,多种复合金属氧化物中存在的协同效应,包括锰镍复合结构[10]、锰铜复合结构[11]、钴锰复合结构[12]、铜钴复合结构[13]和钴镧复合结构[14]等.而Co、Ni、Mn、Cu、 Fe等都是微波高损耗金属,能充分地吸收微波能并将其转换为热能,在微波场中具有良好的升温性能.本课题组前期选用Co-Mn为反应的主体金属氧化物在微波场中催化降解甲苯,发现当Co∶Mn为2∶1时,制备的Mn1Co2金属氧化物表现出最佳的甲苯降解效果和微波吸收性能,微波热场中,甲苯降解率为50%与90%所对应的反应温度比常规电加热场分别降低48 ℃和6 ℃,微波催化氧化在低温条件下表现出更加优异的催化活性[15].但Mn1Co2金属氧化物存在使用寿命较低、高质量流速下催化活性不太理想等问题,为了进一步提升催化剂的稳定性与催化活性,本次研究拟考虑在钴锰复合金属氧化物引入其他金属元素,以改善催化剂的结构性能.镧氧化物因其优异的热稳定性和可变晶相被广泛用作催化剂的添加剂或载体.La2O3可以形成固溶体,可以通过和过渡金属氧化物的相互作用形成ABO3的钙钛矿型结构,例如LaCoO3、LaMnO3.镧的加入有助于改善材料的反应活性、选择性和热稳定性[16],因此,本研究以La为辅助催化剂的添加材料等量替代金属Mn,研究第三类金属La的加入对复合金属结构与催化效果的影响,以期为开发微波催化降解甲苯的高效催化剂提供新路径.
1 材料与方法
1.1 实验试剂
六水合硝酸镧,分析纯,购于天津市科密欧化学试剂有限公司;六水合硝酸钴,分析纯,购于广东光华科技股份有限公司;50%硝酸锰溶液,分析纯,购于天津市光复科技发展有限公司;柠檬酸,分析纯,购于天津市大茂化学试剂厂;氨水,分析纯,购于湖南汇虹试剂有限公司.
1.2 催化剂制备
钴锰镧氧化物采用溶胶-凝胶法合成.以六水合硝酸镧、六水合硝酸钴和50%硝酸锰溶液作为起始原料,以柠檬酸作为螯合剂.硝酸镧、硝酸钴和硝酸锰以一定物质的量比((La+Mn)∶Co=1∶2)溶于适量蒸馏水中,将适量柠檬酸加入溶液中(物质的量比,柠檬酸∶(La+Co+Mn)=1∶1),使用氨水将溶液的pH调节至9~10,得到液体溶胶;将溶胶在80 ℃水浴条件下加热搅拌,直至形成黏稠状的凝胶.将凝胶于160 ℃条件下烘干,继而在600 ℃条件下煅烧6 h,得到目标催化剂.根据制备材料的钴锰镧比例,将物质的量比La∶Mn=1∶0、0.8∶0.2、0.6∶0.4、0.4∶0.6、0.2∶0.8、0∶1的催化剂,分别命名为La1Co2、La0.8Mn0.2Co2、La0.6Mn0.4Co2、La0.4Mn0.6Co2、La0.2Mn0.8Co2、Mn1Co2.
1.3 催化剂表征
采用比表面积分析仪(美国康塔公司,型号NOVA2000e)对材料的比表面积、孔径和孔容进行测试分析,采用X射线多晶粉末衍射仪(日本理学公司,设备型号为D/MAX-Ⅱ-2500/PC)对材料的晶体结构来进行分析,采用扫描电子显微镜(SEM)(Sigma 300,德国ZEISS)观察样品的形貌,采用TEMSOR27型傅里叶变换红外光谱仪(德国布鲁克公司)对材料表面官能团进行分析,采用全自动气体吸附分析仪(美国康塔仪器公司,Autosorb-iQ)测定催化剂的升温还原性能,采用X射线光电子能谱仪(XPS)(K-Alpha,美国Thermo Scientific)分析催化剂表面的元素组成及化学价态.
1.4 实验装置与方法
实验装置如图1所示,催化反应装置分为微波反应器与管式炉反应器,微波反应器通过调节微波功率进行加热,输出最大功率为1 000 W,由热电偶实时监测反应温度,管式炉反应器则为传统的电加热形式.两种反应装置均使用石英管作为反应管,将相应质量的催化剂放入其中,并在石英管中提前充填好一定量的石英棉.
实验方法:用注射泵将液态甲苯注入气化室进行气化,气化后的甲苯(120 ℃)与干燥的空气充分混匀后进入催化反应装置进行反应,尾气经检测后进入盛有磷酸的尾气吸收装置净化、排放.尾气中甲苯浓度采用气相色谱仪检测,CO2浓度采用多功能复合气体分析仪(GT-2000)检测.甲苯活性测试条件为:气体流量为300 mL/min,质量空速(WHSV)为90 000 mL/(g·h),甲苯初始浓度约为2 800 mg/m3.甲苯转化率(η)与矿化率(μ)按下式计算:
(1)
(2)
式中:Co和Ci分别为入口和反应后出口甲苯浓度的数值,单位mg/m3;CCO2,in和CCO2,out分别为甲苯完全氧化时二氧化碳浓度和实时监测反应后出口二氧化碳浓度的数值,单位mg/m3.
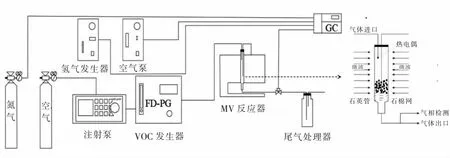
图1 微波催化氧化设备示意图Fig.1 Schematic diagram of microwave catalytic oxidation device
2 结果与讨论
2.1 催化剂表征
2.1.1 X射线晶体衍射(XRD)分析
通过XRD表征分析不同镧掺杂量催化剂的晶体组成和晶体结构,不掺杂镧的Mn1Co2催化剂具有尖晶石结构的MnCo2O4(PDF:23-1237)典型衍射峰(如图2所示).镧掺杂的催化剂,位于2θ为36.82°的(311)衍射峰结晶度减弱,出现了归属LaMnO3(PDF:75-0440)和LaCoO3(PDF:75-0279)的新峰(位于32.61°),催化剂由原本的MnCo2O4尖晶石结构变成了钙钛矿结构的LaMnO3和LaCoO3以及Co3O4,与单一钙钛矿型氧化物相比,LaMnO3和LaCoO3形成的双钙钛矿型氧化物(DPO)可以通过协同效应大大提高金属之间的反应性.不同金属之间的协同作用可以更显著地提高双钙钛矿结构的反应性.同时,由于钴锰离子的半径不同而形成更多的缺陷,有利于形成氧空位,从而提升催化活性[17].
2.1.2 比表面积(BET)分析
从图3可以看出,所有催化剂都表现出IV型等温线,表明催化剂中都存在介孔[18].孔径在16~19 nm之间(如表1所示).各材料均为H3型滞后环,表明材料中的介孔是由颗粒或线状的松散堆积形成的,与SEM分析结果一致.其中,La0.8Mn0.2Co2催化剂具有最大比表面积(69.68 m2/g)和最大孔容(0.13 cm3/g),其值分别是Mn1Co2催化剂的1.64倍和1.60倍,说明金属镧的存在能够增加催化剂中金属氧化物的分散性,增大比表面积.有研究表明,更大的比表面积和孔容,可以提升催化剂吸附甲苯的性能,有利于催化氧化甲苯的进行[19].

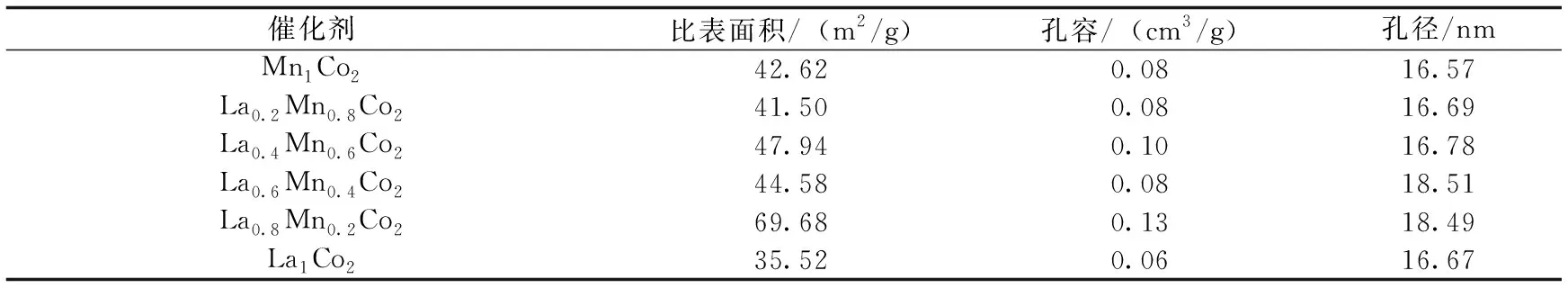
表1 不同材料的比表面积、孔容和孔径
2.1.3 电子扫描电镜表面形貌(SEM)分析
掺杂量的变化能够很大程度上影响材料的微观结构和化学性质,从而影响催化剂的催化性能.由图4可知,3种催化剂表面均匀且多孔,但颗粒形态不均匀,粒径均小于40 nm.与Mn1Co2和La1Co2催化剂相比,La0.8Mn0.2Co2催化剂的颗粒尺寸更小,拥有更多的孔道和较大的比表面积,这可能是由于La0.8Mn0.2Co2催化剂中La的加入,增加了催化剂中金属氧化物的分散性,使催化剂的粒径变小、比表面积增大,这与BET结果一致.此外,相较于La1Co2催化剂,La0.8Mn0.2Co2催化剂拥有更小的粒径,更多的孔道、褶皱和凹陷,这可能是由于LaMnO3和LaCoO3形成的双钙钛矿型氧化物(DPO)中钴锰离子的半径不同,使催化剂表面形成更多缺陷.

图4 催化剂不同放大倍数的SEM图像:(a)Mn1Co2;(b)La0.8Mn0.2Co2;(c)La1Co2Fig.4 SEM images of the catalyst at different magnifications:(a)Mn1Co2 ;(b)La0.8Mn0.2Co2;(c)La1Co2
2.1.4 傅里叶红外光谱(FTIR)
如图5可知,在La掺杂催化剂样品的光谱中观察到位于约457~750 cm-1的特征峰,归因于催化剂中的Mn-O和Co-O四面体的弯曲振动,以1 635 cm-1和3 449 cm-1为中心的谱带观察到表面羟基(-OH)的拉伸振动和羧基和碳基的振动[20].从图谱中可以看出,微波催化氧化甲苯后的催化剂和常规热催化后的催化剂相较于反应前的催化剂,其Mn-O、Co-O基团对应的振动峰均减小.此外,常规热催化后的催化剂,其振动峰小于微波催化后的催化剂,表明在微波催化后的催化剂表面剩余更多的活性基团.Mn-O和Co-O基团的振动峰与催化剂中钴锰所占比例直接相关,占比越高振动峰越大,相对应的催化剂中Mn-O和Co-O基团也越多.
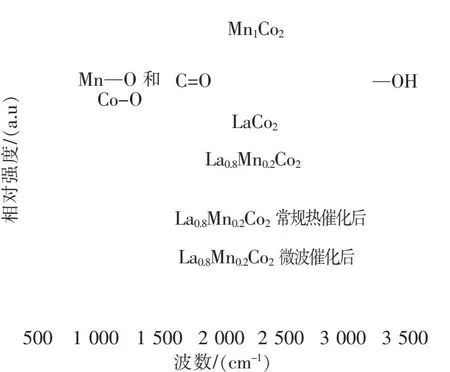
图5 催化剂的FTIR光谱Fig.5 FTIR spectra of the catalyst
2.1.5 X射线光电子能谱(XPS)分析
催化剂的Mn 2p能谱如图6(a)所示.通常金属锰(Mn)以各种氧化物形式存在,包括MnO、Mn2O3、Mn3O4和MnO2.对于Mn1Co2催化剂,Mn2+、Mn3+和Mn4+结合能分别位于640.7 eV、641.8 eV和643.7 eV,Mn2+所占的比例为35.12%,而Mn3+的相对含量为41.04%.从表2可知,相较于Mn1Co2,La0.8Mn0.2Co2的Mn2+占比降低,Mn3+占比达到54.76%,更多的Mn3+物种可以有效提升氧化还原性能[21].且反应前后锰的各价态占比基本没有发生变化,Mn3+和Mn4+相对含量仍然在70%以上.
样品的Co 2p能谱如图6(b)所示,在779.2 eV处的峰归属于Co3+,而在780.9 eV处的峰归属于Co2+,位于782.8 eV和788.8 eV处的2个峰属于振动卫星峰[22].Co3+和Co2+的含量如表2所示,从表中可以看出Mn1Co2和La0.8Mn0.2Co2催化剂中Co3+占比均超过70%,Co3+的占比反映了催化剂的氧化能力[23].从表2中可以看出,反应后Co3+占比均有下降,但是Co3+占比仍超过60%.微波催化后Co3+的占比比常规热催化后的Co3+占比更高,说明微波催化后La0.8Mn0.2Co2催化剂保留更高的反应活性.
样品的O 1s能谱如图6(c)所示,氧的分布如表2所示.对于Mn1Co2催化剂,O 1s峰包含3个重叠峰,分别位于529.5 eV、531.2 eV和532.4 eV,分别对应于晶格氧(表示为Olatt,O22-),表面吸附氧(表示为Oads,O2-或O-)和吸附水中的氧(-OH).对于La掺杂的催化剂,能谱分别以528.3 eV、530.2 eV和531.7 eV为中心的峰.La0.8Mn0.2Co2催化剂中的Oads相较于Mn1Co2明显增加,占比从原来的25.47%增加到了38.63%,其明显增加与催化剂中掺杂镧有关[24].Oads已被证明可促进氧化的活性,可增强氧迁移率[25],有利于催化氧化的进行.同时,O2-和O-等亲电氧物种很容易参与过度氧化和完全氧化,而O2-与选择性氧化有密切关系[26].

图6 Mn1Co2、La0.8Mn0.2Co2、热催化后La0.8Mn0.2Co2催化剂以及微波催化后La0.8Mn0.2Co2催化剂的XPS光谱:(a)Mn 2P;(b)Co 2P;(c)O 1sFig.6 XPS spectra of Mn1Co2,La0.8Mn0.2Co2,La0.8Mn0.2Co2 catalyst after thermal catalysis and La0.8Mn0.2Co2 after microwave catalysis:(a)Mn 2P;(b)Co 2P;(c)O 1s

表2 Mn1Co2、La0.8Mn0.2Co2、热催化后La0.8Mn0.2Co2催化剂以及微波催化后La0.8Mn0.2Co2的XPS参数
2.1.6 H2-程序升温还原(H2-TPR)
采用H2-TPR表征分析了不同材料的还原性能,结果如图7所示,3条还原曲线分别对应Mn1Co2、La0.8Mn0.2Co2和La1Co2催化剂.所有催化剂根据氢气的消耗量均表现出3个还原区域,前两个峰分别对应于Co3+→Co2+→Co0以及MnO2→Mn2O3、Mn3O4→MnO的还原[27].600 ℃左右的小峰对应于钙钛矿尖晶石的还原[28].与Mn1Co2催化剂相比,La掺杂的催化剂的低温还原性明显提高.La0.8Mn0.2Co2催化剂的第一个还原峰温度降至205 °C,第二个还原峰温度降至400 ℃以下,均往低温段移动,有利于氧的迁移转化,表明其还原性能增强,这可归因于表面吸附的氧物质的去除[29].XPS结果也证实催化剂具有更多的表面吸附氧.
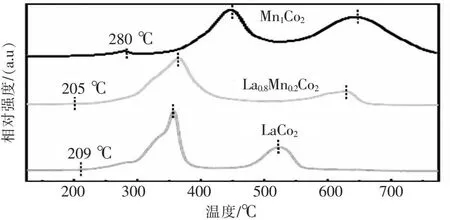
图7 Mn1Co2、La0.8Mn0.2Co2、热催化后La0.8Mn0.2Co2催化剂以及微波催化后La0.8Mn0.2Co2的H2-TPR光谱Fig.7 H2-TPR spectra of Mn1Co2,La0.8Mn0.2Co2,La0.8Mn0.2Co2 catalyst after thermal catalysis and La0.8Mn0.2Co2 after microwave catalysis
2.2 催化剂性能实验
2.2.1 不同镧掺杂量的催化活性与微波升温性能
在甲苯浓度为2 800 mg/m3,WHSV为90 000 mL/(g·h)条件下,对制备的催化剂进行甲苯降解活性测试,反应结果如图8(a)所示.可以发现La0.8Mn0.2Co2催化剂具有最高的催化活性,催化剂在甲苯降解率为50%(T50)与90%(T90)所对应的温度分别为138 ℃和192 ℃.这得益于La0.8Mn0.2Co2催化剂拥有更大的比表面积与最小的粒径,而大的比表面积与小的粒径有利于活性位点的暴露,从而促进催化反应的进行.对比La0.8Mn0.2Co2与La1Co2催化剂可以发现,低温条件下(<130 ℃)La1Co2催化剂催化活性更高,而高温条件下La0.8Mn0.2Co2催化剂催化活性最佳.针对此现象,分别测试了微波功率为300 W时材料的升温性能,如图8(b)所示,可以发现当温度低于120 ℃时La1Co2催化剂在微波场中升温速率更快,表面热点更多,促进催化氧化反应的进行,因此T50所对应的温度最低.当温度达到120 ℃以上时,La0.8Mn0.2Co2催化剂出现热失控现象,表面产生大量热点,迅速升温,大量热点的产生使催化剂催化性能大幅度提升.因此,La0.8Mn0.2Co2催化剂的T90远低于其他催化剂.结果表明,在微波场中催化剂的升温性能也是影响催化性能的重要因素.
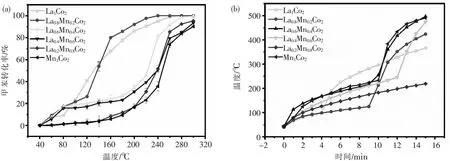
图8 (a)不同镧掺杂量的混合氧化物催化剂催化性能的影响;(b)不同催化剂在微波场中的升温性能测试Fig.8 (a)The effect of different lanthanum doping amounts on the catalytic performance of mixed oxide catalysts; (b)The heating performance test of different catalysts in microwave field
2.2.2 不同质量空速与初始甲苯浓度对材料催化效果的影响
La0.8Mn0.2Co2催化剂在不同质量空速条件下的甲苯降解曲线如图9(a)所示.当WHSV分别在30 000 mL/(g·h)、45 000 mL/(g·h)和90 000 mL/(g·h)时,甲苯各温度段转化率差距不大,T90基本保持在同一水平.当质量空速从90 000 mL/(g·h)增加到120 000 mL/(g·h),可以观察到甲苯的催化活性呈明显的下降趋势,220 ℃条件下的转化率仅剩50%.这是因为高的质量空速缩短了甲苯分子与催化剂上活性位点的接触时间,从而抑制了催化反应.图9(b)为不同甲苯初始浓度条件下的La0.8Mn0.2Co2甲苯降解曲线.可以发现La0.8Mn0.2Co2甲苯的催化活性随着甲苯初始浓度的升高而下降.这是因为催化剂本身活性位点的数量是有限的,当甲苯的浓度远远高于催化剂活性位点的数量时,会使得多余的甲苯分子无法与活性位点接触,从而无法参与催化氧化反应.
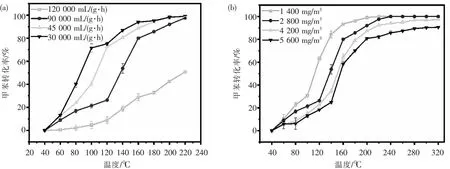
图9 (a)质量空速对催化性能的影响;(b)甲苯初始浓度对催化性能的影响Fig.9 (a)Effect of WHSV on catalytic performance;(b)Effect of initial toluene concentration on catalytic performance
2.2.3 不同热场对甲苯的催化效果和矿化率的影响
图10为不同热场中La0.8Mn0.2Co2的催化活性和矿化率.如图10(a)所示,在常规热场中,催化剂的T50与T90所对应的温度分别为178 ℃和239 ℃,而微波场中催化剂的T50与T90所对应的温度分别为138 ℃和192 ℃,可以发现相同反应温度下,La0.8Mn0.2Co2催化剂在微波场中拥有更高的甲苯催化活性.从图10(b)可以发现,在相同甲苯转化率条件下,La0.8Mn0.2Co2催化剂在微波场中的矿化率远高于常规热场,当甲苯降解率达到50%时,微波加热场与常规电加热场的矿化率分别是40.4%和28.0%,甲苯转化率为90%时所对应的矿化率分别为76.6%和73.8%.通过对比La0.8Mn0.2Co2催化剂常规热催化与微波催化反应前后的Mn-O和Co-O活性基团、吸附氧物种和Co3+物种数量,可以发现波催反应后的催化剂更有利于保留活性物种含量,从而表现出更加优异的催化活性与矿化率.
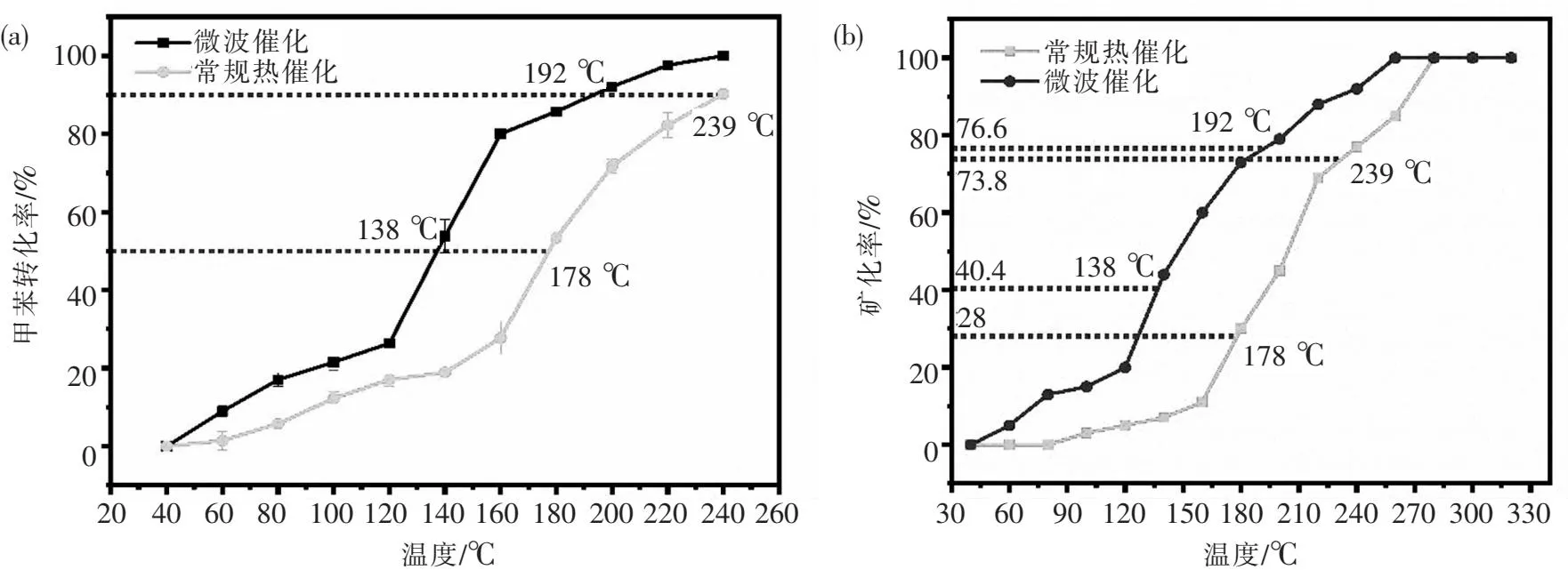
图10 (a)La0.8Mn0.2Co2在不同热场中对甲苯催化性能的影响;(b)La0.8Mn0.2Co2在不同热场中对甲苯矿化率的影响Fig.10 (a)The effect of La0.8Mn0.2Co2 on the catalytic performance of toluene in different thermal fields;(b)The effect of La0.8Mn0.2Co2 on the mineralization rate of toluene in different thermal fields
2.2.4 催化剂寿命测试
为了检验催化剂的稳定性,对La0.8Mn0.2Co2催化剂进行了甲苯催化氧化的连续性测试和重复循环实验.如图11(a)所示,La0.8Mn0.2Co2催化剂在温度为192 °C的条件下连续反应35 h,甲苯转化率基本维持在90%左右.图11(b)为Mn1Co2和La0.8Mn0.2Co2催化剂的重复循环实验,可以发现在前3次的循环实验中,两种催化剂的甲苯转化率均保持在90%左右,从第4次重复实验开始,Mn1Co2催化剂的甲苯转化率逐渐降低,经过10次重复实验后甲苯转化率仅剩75%,而La0.8Mn0.2Co2催化剂的甲苯转化率仍有85%.镧的加入使催化剂拥有更好的分散性,使活性组分受热均匀,这使催化剂不容易因为长时间的加热而失活和团聚,因而表现出了稳定的催化活性,这与BET与SEM测试结果是一致的.
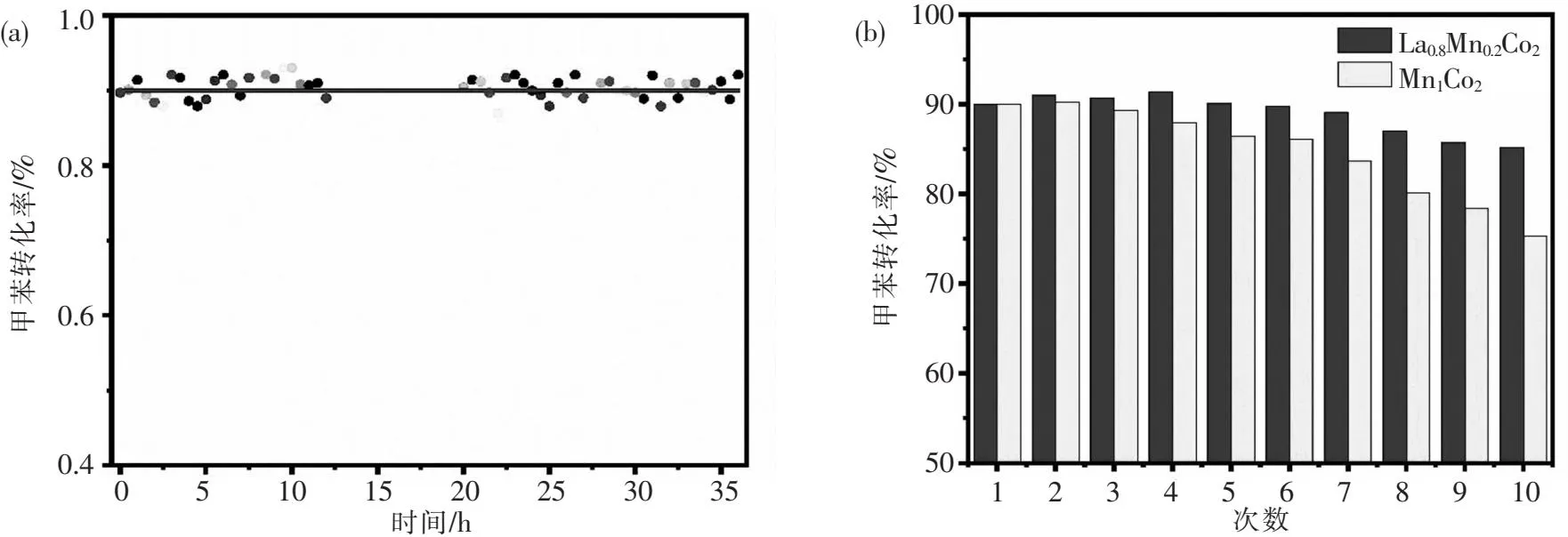
图11 (a)La0.8Mn0.2Co2催化剂在甲苯转化率90%条件下的35 h连续实验;(b)Mn1Co2和La0.8Mn0.2Co2在初始90%甲苯转化率温度下的重复实验Fig.11 (a)35 h continuous experiment of La0.8Mn0.2Co2 mixed oxide catalyst at 90% toluene conversion;(b)Repeated experiment of Mn1Co2 and La0.8Mn0.2Co2 at initial 90% toluene conversion temperature
3 钴锰镧复合金属氧化物催化氧化甲苯的动力学研究
3.1 催化剂对甲苯的反应速率
Alifanti等[30]研究发现,钴锰氧化物作为主要活性成分的催化剂的催化降解甲苯的过程可看作一级反应.为了将内部和外部扩散的影响排除在外,选取甲苯降解率低于20%的4个温度点进行反应速率(ri,mol/(g·h))的计算[31].其中120 ℃、100 ℃、80 ℃和60 ℃4个不同的温度点为微波场的计算温度,180 ℃、160 ℃、140 ℃和120 ℃为常规热场中的计算温度,反应速率的运算方法如公式(3)所示,最终的结果见表3和表4.
(3)
式中:ri为反应速率,单位mol/(g·h);η为特定温度条件下甲苯转化率;Ctoluene为甲苯在气体混合物中浓度的数值,单位mol/mL;F为气体总流量的数值,单位mL/min;m为La0.8Mn0.2Co2催化剂质量的数值,单位g.

表3 微波场中La0.8Mn0.2Co2催化剂在不同温度和不同初始浓度条件下的甲苯降解率及反应速率

表4 常规热场中La0.8Mn0.2Co2催化剂在不同温度和不同初始浓度条件下的甲苯降解率及反应速率

表(4)续
3.2 催化剂的表观活化能
在甲苯浓度为2 800 mg/m3、4 200 mg/m3和5 600 mg/m3条件下,测算微波场中和常规热场中的表观活化能.反应的条件和温度的选择保持与3.2节一致.通过MVK(Mars-van Krevelen)反应动力学模型对整个反应的活化能进行阿伦尼乌斯(Arrhenius)方程拟合来计算表观活化能,如公式(5)和公式(6)所示.从公式(4)中可知,在浓度确定的情况下反应速率r与反应速率常数k成正比.根据公式(7),使用-1/(RT)为横坐标,相应温度下的lnri为纵坐标进行拟合,拟合后直线的斜率就是对应反应的活化能.
ri=kcn,
(4)
(5)
(6)
(7)
式中:k为反应速率常数;c为底物浓度的数值,单位mg/m3;n为反应级数,n=1;A为指前因子(常数);R为摩尔气体常数,单位 kJ/(mol·K);T为反应温度的数值,单位K;Ea为反应活化能的数值,单位kJ/mol.
将表4和表5中的数据带入式(3)~式(5),以-1/(RT)为横坐标,相应温度下的lnri为纵坐标进行拟合,拟合后直线的斜率就是对应反应的活化能,拟合结果如图12所示,从图中可以看出,微波场中,La0.8Mn0.2Co2催化剂在2 800 mg/m3、4 200 mg/m3和5 600 mg/m3甲苯浓度对应的活化能分别是19.21 kJ/mol、26.19 kJ/mol和22.52 kJ/mol.常规热场中,La0.8Mn0.2Co2催化剂在2 800 mg/m3、4 200 mg/m3和5 600 mg/m3甲苯浓度对应的活化能分别是26.13 kJ/mol、30.56 kJ/mol和32.56 kJ/mol.同时所有拟合方程的相关系数R2基本都在0.9以上,说明数据的拟合度较高.可以发现甲苯初始浓度的升高,表观活化能也随之增加,这是由于甲苯浓度的升高会增加催化过程中材料的负担.在微波场中,2 800 mg/m3甲苯初始浓度条件下表现出最低的表观活化能19.22 kJ/mol,相比于常规热场的26.13 kJ/mol降低了6.91 kJ/mol.由此可知,微波场中催化剂降解甲苯所需的活化能得到有效降低,这可能是由于微波场的能量除了使催化剂表面产生热点,为升温提供能量外,还有部分能量通过降低反应的活化能来提高催化剂降解甲苯的活性.
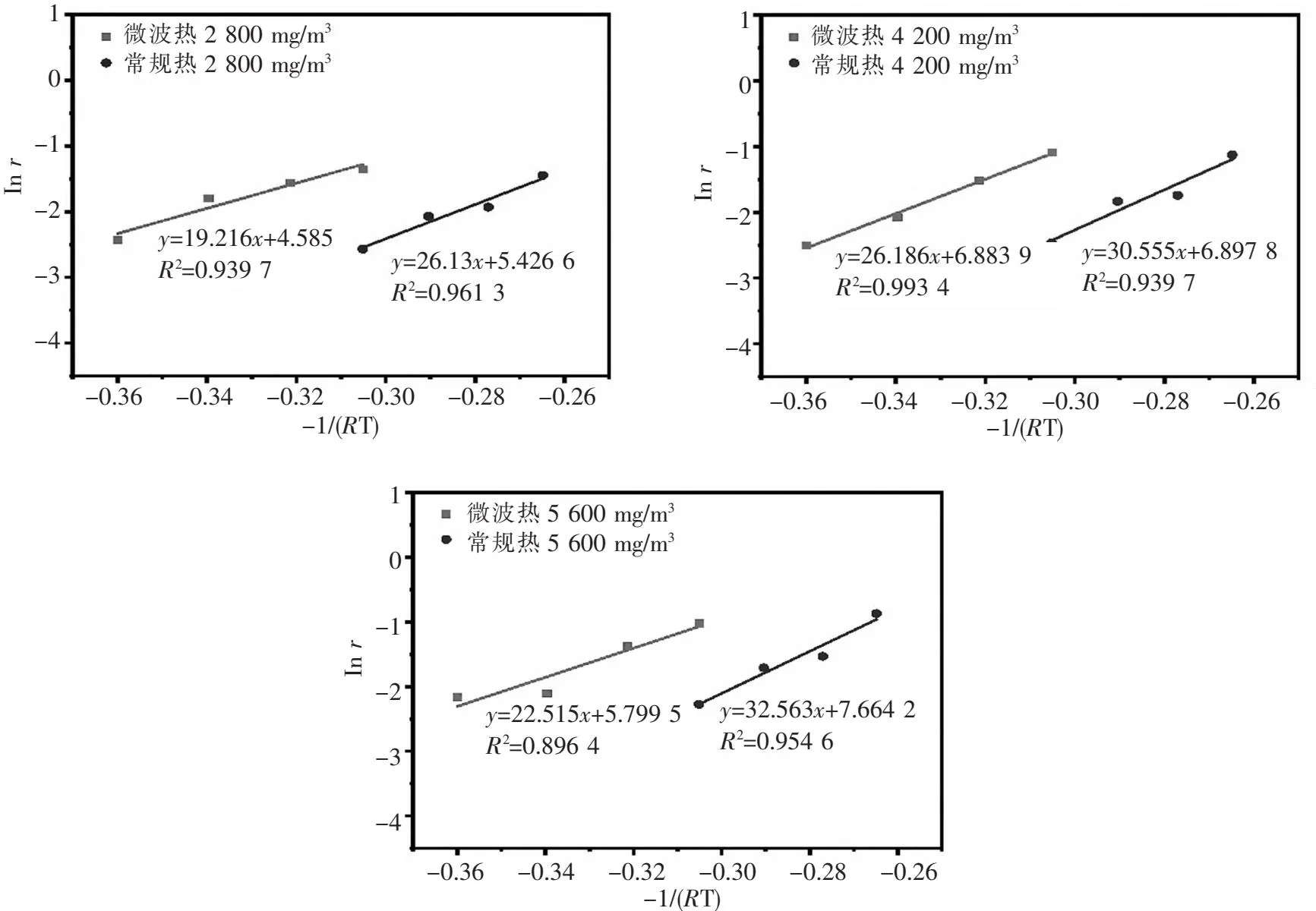
图12 La0.8Mn0.2Co2催化剂在不同热场和不同甲苯初始浓度条件下的Arrhenius拟合图Fig.12 Arrhenius fitting diagram of La0.8Mn0.2Co2 catalyst under different thermal fields and different initial concentrations of toluene
4 结论
在Mn1Co2催化剂中引入第三类金属La,以La为辅助催化剂的添加材料等量替代金属Mn,对Mn1Co2纳米催化剂进行改性.实验表明当La与Mn的物质的量比为4∶1时制备的La0.8Mn0.2Co2催化剂在微波条件下表现出最佳的催化活性,在温度为192 ℃时便可以实现90%的甲苯转化率,经过32 h的连续性测试以及10次的重复性测试仍然能保持良好的催化活性.对La0.8Mn0.2Co2催化剂进行表征与升温测试,表明材料的微波催化氧化活性与稳定性得益于其较大比表面积、更小的粒径、更多的活性基团、丰富的Mn3+、Mn4+和Co3+物种以及优异的微波吸收性能.与常规热催化进行对比,La0.8Mn0.2Co2催化在微波条件下甲苯降解率为50%对应的温度与矿化率分别为138 ℃与40%,优于常规热场中对应的178 ℃与28%.通过对La0.8Mn0.2Co2催化剂进行反应动力学研究,结果表明反应速率与温度和甲苯浓度呈正相关.微波场中在更低的反应温度条件下,就能达到更高的反应速率.在催化温度为120 ℃,甲苯浓度在2 800 mg/m3的条件下,微波场中的反应速率是常规热场的3.36倍.甲苯初始浓度的升高,表观活化能也随之增加.微波场中,当甲苯初始浓度在2 800 mg/m3的条件下表现出最低的表观活化能(19.22 kJ/mol),相比于常规热场的活化能(26.13 kJ/mol)降低了6.91 kJ/mol.综上可知,微波催化能在更低的温度条件下实现更高效率的污染物的降解与转换,由于微波催化独特的加热方式与优异的催化活性,使其催化降解过程更加安全、更加节能,污染物降解更加彻底、更加环保,与常规热催化相比具有更好的应用价值.
