光甘草定/羟丙基-β-环糊精包合物的释放特性、黏液渗透性及细胞摄取
2024-01-03李德锋樊金玲姚培培任国艳张晓宇
李德锋,樊金玲,姚培培,任国艳,杜 琳,张晓宇
(河南科技大学食品与生物工程学院,河南 洛阳 471023)
光甘草定(glabridin,GLD)是光果甘草特有的异黄烷类化合物,具有多种生物活性,如抗炎、抗氧化、抗肿瘤、降血糖、神经保护等[1-5]。但是,GLD的生物利用度很低[6-7],无法在体内充分发挥其功能,从而很大程度上制约了GLD的应用。
溶解度低是GLD 生物利用度低的一个主要原因。小肠上皮细胞主要以被动扩散的跨膜转运方式摄取和转运GLD[7],而被动扩散是由小肠上皮细胞肠腔内消化液中高药物浓度驱动。GLD的水溶性极低(在25 ℃水中溶解度约7 μg/mL[8]),造成口服利用时在肠腔中的浓度很小,严重影响它的小肠吸收率。透过肠上皮细胞的渗透性差是影响GLD 生物利用度的另一重要原因。Ito等[6]报道了10 μmol/LGLD 透过Caco-2 细胞单层模的Papp仅为(1.70±0.16)×10-5cm/s,为低渗透性分子。此外,人体肠道上皮表面存在着由水和黏蛋白为主要成分的黏液层,是影响很多营养成分或药物吸收的重要物理屏障[9]。迄今为止,尚缺少有关GLD对黏液层的渗透性研究。
环糊精(cyclodextrin,CD)具有外亲水、内疏水的独特笼状结构,能够通过静电相互作用、氢键和范德华力等非共价力与多种化合物相互作用,尤其是疏水性化合物,疏水性化合物被包合在CD的空腔中,形成主-客体包合物,从而使难溶性化合物的溶解度得到提高[6,10-12]。研究表明:某些黄酮的CD包合物可促进黄酮化合物的口服吸收,提高其生物利用度[11],可能途径及机制有:1)改善难溶性黄酮化合物的溶出动力学,提高其瞬时饱和溶解度[10];2)增强亲脂性黄酮化合物透过黏液层的能力,提高肠壁药物浓度[12];3)增加黄酮化合物的细胞膜渗透性等[13]。
本团队的前期研究表明:多种CD 对GLD 均有很好的包合作用[14]。本实验选用羟丙基-β-环糊精(hydroxypropyl-β-cyclodextrin,HP-β-CD)包合GLD,采用冷冻干燥法制备了GLD/HP-β-CD包合物,旨在提高GLD在水中的溶解度;通过扫描电镜(scanning electron microscopy,SEM)、差示扫描量热法(differential scanning calorimetry,DSC)、傅里叶变换红外光谱(Fourier transform infrared spectroscopy,FTIR)法和分子对接技术分别对包合物的表面状态、GLD的存在形式、GLD与HP-β-CD的相互作用及空间构象进行了分析;采用体外实验研究GLD被包合前后在胃、肠液中的溶出和释放特性,利用Transwell小室法研究了包合前后GLD在黏液层中的渗透性;采用Caco-2细胞研究了包合对小肠摄取GLD的影响。研究探讨载体HP-β-CD对GLD吸收的影响及可能机制,以期为进一步推动GLD在功能食品、膳食补充剂等领域中的应用提供实验依据。
1 材料与方法
1.1 材料与试剂
GLD 洛阳蓝斯利科技有限公司;0.45 μm滤膜日本Millipore公司;HP-β-CD(6位羟基取代,取代度为5.8~7.5)湖北恒硕化工有限公司;MTT上海源叶生物科技有限公司;无水乙醇、石油醚、氯化钠、乙腈、盐酸、氢氧化钠、磷酸二氢钾、冰醋酸、二甲基亚砜、吐温-80 天津市德恩化学试剂有限公司;溴化钾 上海网化化工科技有限公司;Transwell-24孔板 美国康宁有限公司;铝坩埚 上海菁仪化工材料有限公司;再生纤维素透析袋(800~1 000 Da)上海吉至生化科技有限公司;黏液素II 上海臻科生物科技有限公司;BCA蛋白质试剂盒 上海钰博生物科技有限公司;MEM培养基、双抗 美国Hyclone公司;胎牛血清 美国Gibco公司;胰酶(含酚红)德国Merck公司;Caco-2细胞 美国典型培养物保藏中心。
1.2 仪器与设备
L5S紫外分光光度计 浙江恒岳仪器有限公司;TENSPOR27 FTIR光谱仪 德国Bruker公司;SCIENTZ-10N冷冻干燥机 西安蔚然电子科技有限公司;DSC1型DSC仪上海胥元机械有限公司;EClassical 3100高效液相色谱(high performance liquid chromatography,HPLC)仪苏州格雷弗工业装备有限公司;SEM 北京铂瑞达科技有限公司;DF-101S水浴加热磁力搅拌器 济南欧莱博生物科技有限公司;KQ-500DE型数控超声波清洗器杭州微米派科技有限公司;SW-CJ-2FD双人单面超净工作台 苏州净化设备有限公司;CQT-191IR恒温培养箱 上海金畔生物科技有限公司;RS-232C酶标仪美国Bio-Rad公司。
1.3 方法
1.3.1 GLD/HP-β-CD包合物的制备
按物质的量比1∶1分别称取适量的GLD和HP-β-CD,然后用乙醇将其配成质量浓度为30 mg/mL的GLD溶液;用去离子水将其配成浓度为0.04 mol/L的HP-β-CD溶液。将两种溶液混合,并在25 ℃、200 r/min条件下持续振荡24 h。在旋转蒸发器(45 ℃)中除去所得溶液中所含有的乙醇,之后在-20 ℃的环境中预冻12 h。最后在冷冻干燥机中冻干,将冻干后的样品过80 目筛,即得到所需包合物。
1.3.2 GLD/HP-β-CD包合物的饱和溶解度、包合率和载药率的测定
1.3.2.1 饱和溶解度
在室温条件下(25 ℃)借助超声,使GLD/HP-β-CD包合物在1 mL水中溶解至平衡状态。溶液过0.45 μm滤膜后,其滤液用80%乙醇溶液进行稀释。在281 nm波长处测定吸光度,并根据得到的吸光度计算溶液中GLD的含量,即为饱和溶解度。
1.3.2.2 包合率和载药率
参照文献[15-17]。分别称取两份质量均为10 mg的GLD/HP-β-CD包合物。其中一份先用1 mL去离子水溶解,加入9 mL乙腈,将其在25 ℃条件下,经超声处理20 min。5 000 r/min离心5 min后,将上清液收集到新离心管中,用乙腈稀释10 倍。在281 nm波长处测定其吸光度,根据标准曲线计算出溶液中GLD的质量,即为包合物样品中GLD的总质量。另一份包合物用石油醚(400 μL)溶解,离心后弃掉上清液(除去未被包合的GLD);重复操作两次;在通风橱中,使样品中的石油醚挥发;按上述包合物样品中GLD总质量的测定步骤对此样品中的GLD质量进行测定,即被包合的GLD质量。按照下式计算包合率和载药率:
1.3.3 GLD/HP-β-CD固体包合物的结构表征
1.3.3.1 SEM
将GLD、HP-β-CD、GLD与HP-β-CD的物理混合物(以物质的量比1∶1均匀混合)及GLD/HP-β-CD包合物用导电的双面胶固定在样品台上,并喷镀铂金,在3 kW条件下观察各样品的表面形态。
1.3.3.2 DSC分析
分别称取5 mg的GLD、HP-β-CD、GLD与HP-β-CD的物理混合物(以物质的量比1∶1均匀混合)及GLD/HPβ-CD包合物进行DSC的测定。其氮气流量为10 mL/min,温度扫描的范围设定在25~300 ℃,升温速率设为10 ℃/min,并以空盘作空白对比,记录每个样品的DSC曲线。
1.3.3.3 FTIR光谱法
将适量的GLD、HP-β-CD、GLD与HP-β-CD的物理混合物(以物质的量比1∶1均匀混合)及GLD/HP-β-CD包合物分别与溴化钾粉末以质量比1∶100均匀混合,然后在研钵中充分研磨,压片制得样品薄片。利用FTIR检测样品时,背景单通道扫描的样品是纯溴化钾粉末。扫描范围设定为400~4 000 cm-1,分辨率为4 cm-1,扫描32 次。
1.3.3.4 分子对接
β-CD模型文件在剑桥晶体数据库(https://ccdc.cam.ac.uk/)中下载,其编号为1107194,去H2O后,利用Gauss View软件将7 个葡萄糖单元中的6 位羟基氢用羟丙基进行替换,取代度为7;再通过Gaussian 09软件中半经验算法的PM6基组,对几何构型进行优化,得到HP-β-CD模型;GLD 3D模型文件在PubChem数据库(https://pubchem.ncbi.nlm.nih.gov/)中下载,其CID编号为124052,利用Gaussian 09软件中的DFT方法(B3LYP/6-31G)对3D模型进行优化,再通过Open Babel GUI软件将优化后的文件格式转化为pdb格式。
采用Auto Dock Tools 4.2软件对受体(HP-β-CD)与配体(GLD)分别处理,添加原子电荷和H原子,并在GLD分子内设置可旋转单键数量及根原子,处理好的受体和配体保存为pdbqt文件格式。对接方法采用半柔性对接。分子对接时,以对接盒子的中心点为受体几何中心,并且盒子的尺寸设置为60 Å×60 Å×60 Å。搜索参数选用拉马克遗传基因算法、算法对接轮数设为100、能量评估的最大数目设为250 000,其他参数取默认值。
1.3.4 GLD/HP-β-CD包合物在胃肠液中的溶出
β-CD在胃及小肠部分不被人体消化酶作用[18]。按照Maltais等[19]的方法配制不含消化酶的模拟胃、肠液。用100 mL去离子水将准确称取的NaCl(2.0 g)溶解在烧杯中,用浓盐酸调节pH值至1.2,即为模拟胃液。用250 mL去离子水将准确称取的KH2PO4(6.8 g)也溶解在烧杯中,用0.2 mol/L NaOH溶液调节pH值至6.9,即为模拟肠液。量取出900 mL胃液或肠液,将其放置于1 000 mL烧杯内,作为溶出介质。在水浴加热方式的磁力搅拌器中进行搅拌,其水温保持(37±0.5)℃,搅拌的转速保持50 r/min。称取一定量的GLD、GLD与HP-β-CD的物理混合物及GLD/HP-β-CD样品(GLD含量为100 mg),分别加入烧杯中,并立即开始计时;分别在2、4、6、8、10、15、20、30、45、60 min时吸取1 mL溶出液,同时添加等量新鲜的溶出介质。取出的样液过0.45 μm的微孔滤膜后,取0.5 mL的滤液,并向其中加入4.5 mL乙腈,充分混匀后,在5 000 r/min离心5 min,取上清液,在281 nm波长处测定吸光度,按下式计算溶出率:
式中:A为某一时间点每1 mL 样液中溶出的GLD质量/μg;B为之前所有时间点取出的1 mL液体中GLD质量/μg。
1.3.5 GLD/HP-β-CD包合物在胃肠液中的释放
1.3.5.1 透析袋预处理
将透析袋(800~1 000 Da)剪成8~10 cm小段,去离子水清洗后方可使用。
1.3.5.2 释放介质的配制
按照1.3.4节所述配制模拟胃、肠液。在模拟胃、肠液中分别加入一定量吐温-80(体积分数2%)作为胃、肠液释放介质,其中吐温-80用于促进GLD在介质中溶解。
1.3.5.3 HPLC法测定GLD含量
采用EClassical 3100 HPLC系统测定GLD的含量;色谱条件:色谱柱ZORBAX SB-C18(4.6 mm×250 nm,1.7 μm);流动相:A为水(含1%冰醋酸),B为乙腈,采用A∶B=40∶60进行等度洗脱;柱温30 ℃;流速1 mL/min;进样量20 μL;检测波长281 nm。GLD 质量浓度0~100 ng/mL,标准曲线方程为y=0.164 92x+0.117 58。
1.3.5.4 释放实验
烧杯中加300 mL胃液释放介质(含2%吐温-80的模拟胃液);精确称取一定量GLD/HP-β-CD样品,将其溶于去离子水中,使GLD质量浓度均为2 mg/mL。分别取3 mL样品液放入预处理过的透析袋中,用夹子夹紧两端,放入烧杯中,保证透析袋完全浸没在释放介质中。将烧杯放入37 ℃水浴锅中,每间隔一段时间(0.5、1、2 h)从烧杯中取出1 mL袋外溶液,同时补充1 mL新鲜的胃液释放介质。2 h后,将烧杯中的释放介质更换为肠液释放介质,同样每间隔一段时间(4、7、16、20、24 h),从烧杯中取出1 mL袋外溶液,同时补充1 mL新鲜的肠液释放介质。
将上述不同时间段取出的1 mL释放介质于冷冻干燥机中冻干。加入0.2 mL乙腈进行复溶,0.45 μm滤膜过滤,采用HPLC测定GLD的浓度(色谱条件见1.3.5.3节)。计算1 mL释放介质中GLD的质量,并按下式计算累积释放率:
式中:A为某一时间点每1 mL样液中释放的GLD质量/μg;B为之前所有时间点取出的1 mL液体中释放出的GLD质量/μg。
1.3.6 GLD/HP-β-CD包合物在黏液层的渗透性
1.3.6.1 黏液的配制
配制pH 7.4的缓冲溶液,然后称取一定量的黏蛋白,将其溶接在pH 7.4的缓冲溶液中,配成一定浓度的黏蛋白溶液。
1.3.6.2 黏液渗透实验(Transwell小室扩散法)
利用24 孔Transwell小室研究不同GLD-样品在黏液层中的扩散行为,该装置包含一个供体室和受体室,同时中间有个聚碳酸酯半透膜,它能允许粒子穿过但黏液不能渗透通过。Transwell小室在使用前,供体室和受体室中均加入pH 7.4的磷酸缓冲液,平衡30 min。将40 μL的黏蛋白溶液加入到供体室的半透膜上,受体室加入1 000 μL的磷酸缓冲液,上室分别加入200 μL相同质量浓度的1 mg/mL的GLD/DMSO或者GLD/HP-β-CD溶液,将装置至于37 ℃转速为100 r/min的摇床上培养6 h,于1、2、3、4、5、6 h时,从受体室取出0.15 mL,并向室中加入等量的预热缓冲液。
1.3.6.3 GLD含量测定
GLD/DMSO组的样品取出后于冷冻干燥机中冻干,后加入200 μL乙腈复溶,0.45 μm滤膜过滤,采用HPLC法检测其中GLD的含量,HPLC法的条件见1.3.5.3节;GLD/HP-β-CD组的样品取出后可直接用紫外分光光度计检测其吸光度并计算其中GLD的含量。
1.3.6.4 表观渗透系数
黏液渗透结果用表观渗透系数(Papp)表示,计算公式如下:
式中:dQ/dt为受体室在单位时间获得的药物量/(μg/(mL·s));A为Transwell的膜表面积/cm2;C0为药物的初始质量浓度/(μg/mL)。
1.3.6.5 GLD与黏蛋白的分子对接
应用AutoDock Tools 4.2软件将黏蛋白(MUC2)与GLD进行分子对接。GLD分子模型的建立参照1.3.3.4节;从PDB数据库中得到了单体MUC2(MUC2-1)(PDB ID:6TM6)和四聚体MUC2(MUC2-4)(PDB ID:6RBF),采用Auto Dock Tools 4.2工具对上述蛋白受体和配体进行常规处理,再用其Autogrid模块得到对接活性位点,然后开始进行分子对接,得到结合能,探讨GLD与两种MUC2之间的亲和力。并利用PyMOL和Discovery Studio 4.5软件对分子对接进行可视化分析。
1.3.7 Caco-2细胞对GLD的摄取
1.3.7.1 样品准备
用DMSO溶解GLD,并将其配制成32.44 mg/mL的母液,然后用MEM培养基稀释为不同浓度的样品(DMSO体积分数0.1%),其浓度分别为10、30、50、70、90、100 μmol/L,记为GLD/DMSO组;用MEM培养基溶解GLD,并配制成浓度梯度同GLD/DMSO组的溶液,记为GLD/H2O组。用MEM培养基复溶GLD/HP-β-CD包合物,并配制成浓度梯度也同GLD/DMSO组的溶液,记为GLD/HP-β-CD组。
1.3.7.2 样品对Caco-2细胞的细胞毒作用
采用MTT法测定不同浓度样品对Caco-2细胞增殖的抑制作用,用于确定Caco-2细胞摄取实验中GLD的安全使用浓度。在37 ℃、5% CO2气体的恒温培养箱中,将Caco-2细胞在含20%胎牛血清、100 U/mL双抗的MEM培养基中培养至80%(铺满瓶底)。用胰酶消化8 min后,调整细胞浓度为1×105个/mL,将其接种在96 孔板中,每孔200 μL,培养24 h。弃去旧培养基,加入200 μL新鲜培养基或样品液,作用4 h。结束后,用磷酸盐缓冲液(phosphate buffered saline,PBS)清洗2 次,每孔加入200 μL的新鲜培养基和10 μL的MTT,继续作用4 h。弃去培养基,添加150 μL DMSO溶液,10 min后,用酶标仪于波长550 nm处测定各孔吸光度。未添加MTT的孔记为A0,添加等体积培养基的孔记为A1,添加MTT的样品孔记为A2,每个样品6 个平行孔,根据下式计算细胞存活率:
1.3.7.3 细胞摄取实验
取对数生长期的Caco-2细胞进行消化,计数后以2.5×105个/mL接种在6 孔板内,每孔加入2 mL细胞悬液,在37 ℃、5% CO2气体的恒温培养箱中培养;待细胞融合至80%~90%后(铺满瓶底),去除旧培养基,分别加入50 μmol/L的样品液,每孔2 mL。培养4 h后吸出培养基,用PBS轻轻洗涤2 次(以下步骤均在4 ℃条件下进行),加入0.5 mL胰酶消化8 min,加1 mL MEM培养基中止消化;收集细胞至2 mL的离心管内,1 500 r/min离心3 min,去除上清液;加入1 mL预冷的去离子水重悬细胞,超声粉碎(功率350 W,工作3 s,停10 s,工作30 次,超声仪提前用冰块降温至4 ℃);随后高速冷冻离心机离心(10 000 r/min,10 min,4 ℃),吸取上清液0.9 mL用于后续实验。
其中,先取上述0.9 mL上清液中的0.8 mL放离心管中于冷冻干燥机中冻干。冻干后,GLD/H2O、GLD/DMSO组样品加入0.2 mL乙腈进行复溶;GLD/HP-β-CD组样品先加入20 μL去离子水,后加入0.18 mL乙腈进行复溶;复溶后样品用0.45 μm滤膜过滤,采用HPLC法测定上清液中的GLD浓度(色谱条件见1.3.5.3节)。
同时,再取上述0.9 mL上清液中的20 μL,采用4,4′-二羧基-2,2-联喹啉法(bicinchoninic acid,BCA法)进行细胞蛋白含量的测定。
1.3.7.4 BCA法测定细胞蛋白含量
标准曲线的绘制:将蛋白质标准品(1 mg/mL)按0、20、40、60、80、120、160、200 μL的梯度分别加入到1.5 mL离心管中,添加标准品稀释液,使最终每管体积为200 μL,且标准品的质量浓度依次为0、0.1、0.2、0.3、0.4、0.6、0.8、1.0 mg/mL。将上述标准品按其质量浓度梯度依次加入96 孔板中,每孔20 μL,且每孔3 个平行,再向各孔中加入200 μL的BCA溶液,在37 ℃烘箱中反应30 min,最后在酶标仪562 nm测定每孔的吸光度。绘制标准曲线,其横坐标为蛋白质质量浓度,纵坐标为吸光度。标准曲线方程为y=0.614 7x+0.013 3。
细胞蛋白含量的测定:在96 孔板中加入样品,每孔20 μL,每个样品3 个平行孔,每孔再加入200 μL的BCA溶液,在37 ℃烘箱中反应30 min,在562 nm波长处测定各孔的吸光度。根据标准曲线可得细胞中蛋白质的含量。
1.3.7.5 细胞摄取量计算
根据下式计算细胞的摄取量:
式中:CGLD为细胞内GLD的质量浓度/(μg/mL);Cprotein为细胞悬液中蛋白质量浓度/(mg/mL)。
1.4 数据统计及图像处理
2 结果与分析
2.1 GLD/HP-β-CD的制备及结构表征
2.1.1 GLD/HP-β-CD的制备
经冷冻干燥法制备的GLD/HP-β-CD包合物如图1所示,GLD/HP-β-CD的包合率、载药率和饱和溶解度见表1,其包合率和载药率分别为90.03%、14.51%,载体HP-β-CD能使GLD在水中的饱和溶解度显著提高至109.36 mg/mL。Wei Yongqin等[8]研究表明GLD在25 ℃水中溶解度约7 μg/mL。与此相比,GLD经HP-β-CD包合后其在水中的溶解度提高了15 622 倍。已有研究表明,通过将β-乳球蛋白[20]、卵清蛋白[21]分别与GLD形成复合物后,可以使GLD在水中的溶解度分别提高至0.231 mg/mL和0.032 mg/mL。而采用HP-β-CD包合GLD,使其在水中的溶解度远高于上述研究中的方式。

表1 GLD/HP-β-CD的包合率、载药率和饱和溶解度Table 1 Encapsulation efficiency,drug loading and saturation solubility of GLD/HP-β-CD

图1 GLD/HP-β-CD固体包合物Fig.1 Visual appearance of GLD/HP-β-CD solid inclusion complexes
2.1.2 GLD/HP-β-CD的结构表征
2.1.2.1 SEM
利用SEM观察到GLD、HP-β-CD、GLD与HP-β-CD的物理混合物及GLD/HP-β-CD包合物的外部形貌如图2所示,GLD呈柱状晶体结构,轮廓清晰;HP-β-CD呈表面多孔的球形结构[22];物理混合物中同时观察到柱状的GLD和球状HP-β-CD;GLD/HP-β-CD固体包合物呈边缘锋利、形状不规则的片状结构。

图2 GLD/HP-β-CD的SEM图像Fig.2 SEM image of GLD/HP-β-CD
2.1.2.2 DSC
如图3所示,在233 ℃处,发现到GLD有尖锐的熔融峰,此温度是该晶体的熔点[23]。在94.08 ℃处,显示出HP-β-CD具有较宽的吸热熔融峰,这与HP-β-CD水分损失有关[24]。在物理混合物的DSC曲线中发现,其曲线图是GLD与HP-β-CD吸热峰的简单叠加,表明这两种物质仅是简单的物理混合而彼此之间未发生相互作用,在物理混合物中GLD仍表现出晶体特征。在GLD/HP-β-CD包合物的DSC曲线中,HP-β-CD在94.08 ℃处的峰偏移至77.62 ℃,并且强度降低,GLD特征吸热峰则完全消失,这可能是由于药物分子取代了HP-β-CD空腔内的水分子造成[25]。以上结果表明,在GLD/HP-β-CD包合物中,GLD是以无定形非晶体的形式存在,GLD已被HP-β-CD包合在空腔内形成了包合物。

图3 GLD/HP-β-CD的DSC分析Fig.3 DSC analysis of GLD/HP-β-CD
2.1.2.3 FTIR光谱
如图4所示,在GLD的光谱中,峰位3 346 cm-1出现一强而宽的峰,为O—H的伸缩振动,1 518、1 464 cm-1为芳香环C=C的伸缩振动,2 964、2 920 cm-1为—CH3的吸收峰[20]。HP-β-CD红外光谱图,在波数为3 401 cm-1处有一个由O—H 官能团的振动而产生的宽峰,在2 930 cm-1处有一个峰,由O—CH3官能团的振动产生;1 156、1 083、1 032 cm-1处各有一个峰,是C—O的振动吸收峰[26]。在物理混合物的光谱中,GLD和HP-β-CD的特征吸收峰依然存在,表明物理混合物只是主客体分子的简单叠加,GLD和HP-β-CD之间没有相互作用或仅有微弱的相互作用。GLD/HP-β-CD包合物在波数为3 385 cm-1处有一个宽峰,该峰应由HP-β-CD的3 401 cm-1偏移所至,这可能是由GLD与HP-β-CD分子间氢键的相互作用造成[27];与物理混合物光谱相比,GLD/HP-β-CD包合物在波数1 518 cm-1和1 464 cm-1处的吸收峰明显减弱,这可能是由于GLD进入到HP-β-CD分子的空腔中从而使其芳香环C=C的伸缩振动减弱。
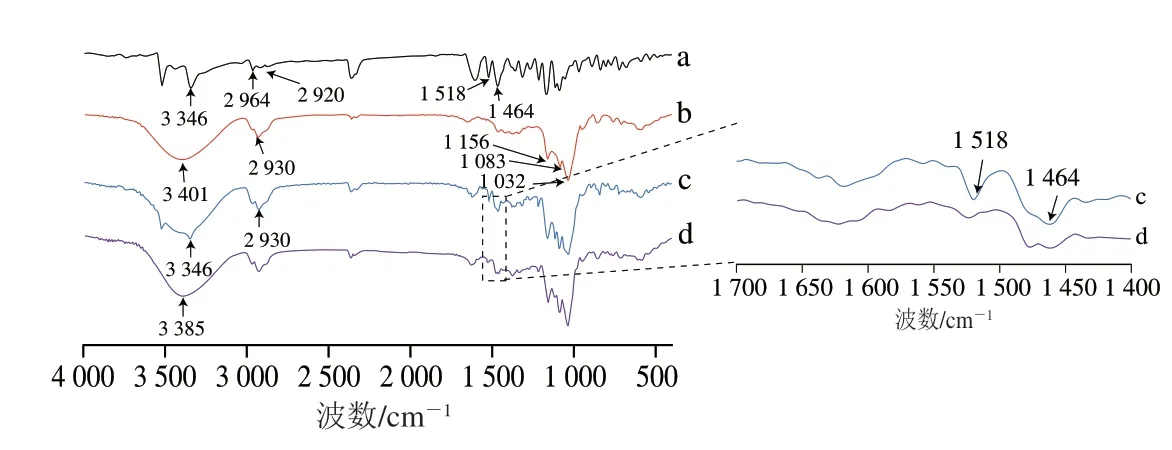
图4 GLD/HP-β-CD的FTIR分析Fig.4 FTIR analysis of GLD/HP-β-CD
2.1.2.4 分子对接
如图5所示,GLD分子能够完整地进入到HP-β-CD的空腔内,以A环指向CDs的小口端为最佳构象;B环上C2位的OH氢与HP-β-CD某2 个葡萄糖单元中间的氧之间形成一个氢键(键长为2.0 Å),氢键的形成有助于包合物的稳定。GLD与HP-β-CD之间的最佳结合能为-7.37 kcal/mol,分子间的范德华力为-8.12 kcal/mol,表明GLD与HP-β-CD分子间相互作用的主要作用力是范德华力。
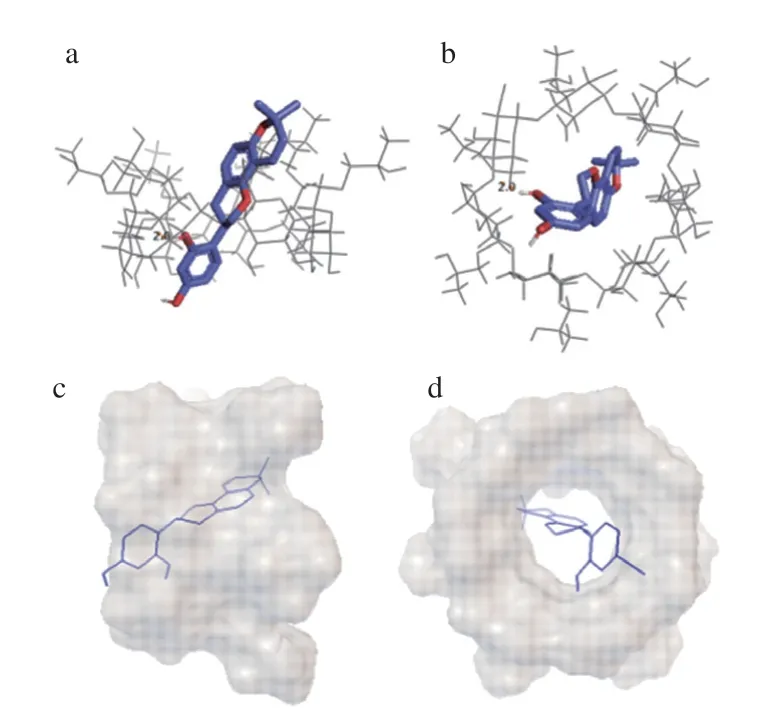
图5 GLD与HP-β-CD对接的最佳能量构象及相互作用图Fig.5 Optimal energy conformation and interaction of GLD with HP-β-CD investigated by molecular docking
2.2 GLD/HP-β-CD提高GLD的小肠吸收
2.2.1 GLD/HP-β-CD在模拟胃、肠液的溶出特性
难溶性的药物或生物活性成分的累积溶出率较低,是导致其在体内吸收率低、生物利用度差的重要原因之一[28]。因此,可以通过累积溶出率在一定程度预测难溶性化合物在体内的吸收速率和利用程度[29]。本实验采用体外模拟实验,以模拟胃、肠液为溶出介质,将透过0.45 μm微孔滤膜的样品计为溶出样品,通过定时取样测定溶出样品中的GLD含量,计算累积溶出率、绘制溶出曲线,研究HP-β-CD包合对GLD溶出特性的影响,结果如图6所示。在模拟胃液和肠液中,GLD在1 h的累积溶出率分别为4%和5%;物理混合物的溶出率略有提高,1 h时溶出率分别约为9%和8%;GLD/HP-β-CD包合物的溶出率均极显著提高,1 h时溶出率分别为63%和62%,是未被HP-β-CD包合GLD累积溶出率的15.75、12.4 倍。表明GLD经HP-β-CD包合后不仅水溶性显著提高,在胃、肠液中的溶出速率和累积溶出率也极显著改善,这是由于GLD与CD上的氢键相互作用以及GLD在包合物中的结晶度减少引起。
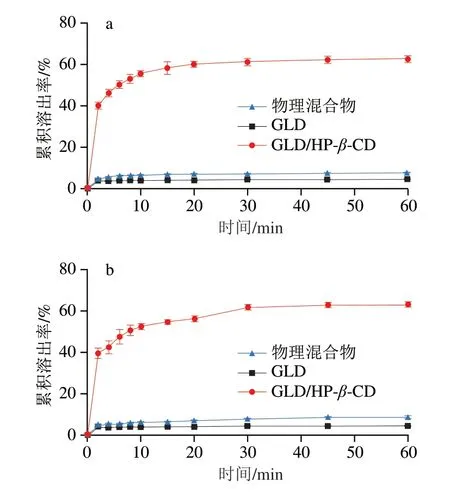
图6 GLD/HP-β-CD在模拟胃液(a)、肠液(b)中的溶出曲线Fig.6 Dissolution curves of GLD/HP-β-CD in simulated gastric (a) and intestinal fluid (b)
2.2.2 GLD/HP-β-CD在模拟胃、肠液的释放特性
药物或活性分子从CD载体中游离出来的释放过程与CD载体药物的溶解过程同时进行。CD与药物的结合力很大程度上决定了释放量,也由此影响了药物吸收:二者结合越强,药物或活性分子越难从载体中释放,吸收就越少。因此,CD包合通常情况下总能增加药物的溶解度、改善溶出性质,但吸收增加不是必然结果。因此,越来越多致力于提高药物吸收的研究更加关注药物的释放过程。
GLD具有溶解度低、肠道渗透性差的特点。根据BCS分类,为BCS IV类药物。提高GLD的水溶性、改善其渗透性,是提高其在体内吸收利用的关键。本部分采用截留分子质量为800~1 000 Da的透析袋体外实验方法研究GLD和GLD/HP-β-CD包合物中的GLD在胃液2 h、肠液22 h连续条件下的释放过程(由于实验初期GLD的释放量较小,本部分采用HPLC法检测透过液中的GLD含量),结果如图7所示。GLD在20 h时开始检测到GLD,24 h的累积释放率为0.32%;GLD与HP-β-CD物理混合物在第16小时开始检测到GLD,第24小时的累积释放率为1.33%;GLD/HP-β-CD从1 h开始检测到GLD,在此后的24 h内累积释放率呈现线性增长趋势,第24小时达16.98%,相对未被包合GLD累积释放率提高约53 倍。这一结果表明:HP-β-CD包合不仅显著提高了的GLD溶解度、改善了溶出性质,同时包合物中的GLD在肠液中能较好地释放,预示着GLD/HP-β-CD可有效提高肠腔中游离GLD分子的浓度。
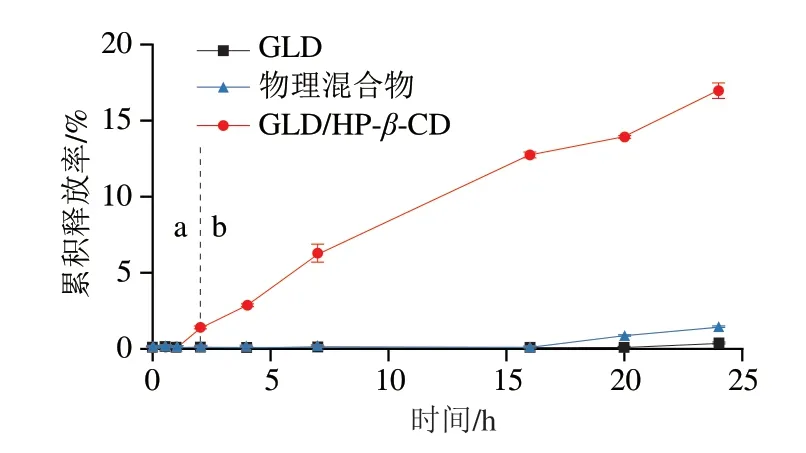
图7 GLD/HP-β-CD在胃肠液连续条件下的释放曲线Fig.7 Continuous release curve of GLD/HP-β-CD in simulated gastrointestinal fluid
2.2.3 GLD/HP-β-CD在黏液层的渗透性
人体肠道上皮表面存在着一层由水和黏蛋白为主要成分的黏液;黏液不断地从黏膜表面分泌和脱落,形成一种立体网格状、且动态变化的物理屏障,具有清除有害物质的作用,但同时也限制了药物的吸收[9]。本实验利用Transwell小室法研究了HP-β-CD包合前后GLD透过黏液层的能力。为考察在溶解状态下游离的GLD分子渗透通过黏液层的能力,本实验将GLD充分溶解于DMSO后再进行Transwell小室的透过实验,以排除GLD不溶于水时聚集成大颗粒对实验结果的干扰。结果表明:采用HPLC法,第4小时接受室开始检测到GLD,对应的累积渗透率仅为0.02%、渗透系数仅为9.24×10-9cm/s,说明游离的GLD分子几乎无法渗透黏液层;GLD/HPβ-CD在黏液层中4 h的累积渗透率为32%,渗透系数为1.43×10-5cm/s,是GLD的1 600 倍(表2)。实验进一步选用不同质量浓度黏蛋白进行GLD/HP-β-CD的渗透实验,结果显示:黏蛋白质量浓度越大,GLD/HP-β-CD累积渗透率越小(图8)。上述研究表明:黏蛋白构成的黏液层是GLD到达肠上皮细胞的主要屏障,而GLD/HP-β-CD则显著提高了GLD透过黏液层的能力。
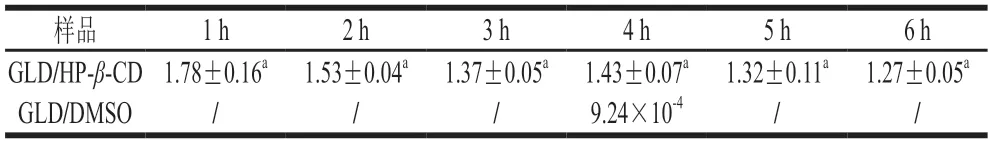
表2 不同样品在20 mg/mL黏液层中Papp值Table 2 Papp values of GLD/HP-β-CD in 20 mg/mL mucous layer 10-5 cm/s
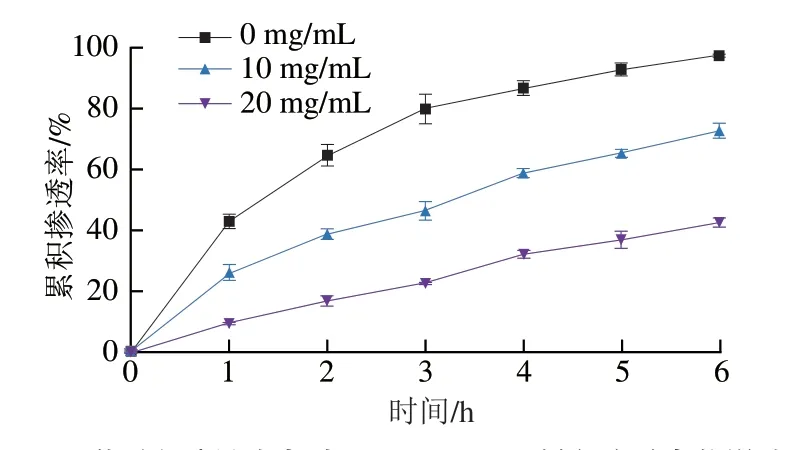
图8 黏蛋白质量浓度对GLD/HP-β-CD累积渗透率的影响Fig.8 Effects of different concentrations of mucin on the cumulative permeability of GLD/HP-β-CD
为进一步探讨GLD透过黏液层能力差的原因,采用分子对接技术研究GLD与肠道黏液层的主要成分MUC2的单体(MUC2-1)和四聚体(MUC2-4)的相互作用,结果图9所示。GLD与MUC2-1的结合位点位于MUC2-1的表面凹陷中,与MUC2-4的结合位点位于4 个MUC2-1相互接合区域的凹槽中;与MUC2-1和MUC2-4的结合能分别为-6.95 kcal/mol和-5.85 kcal/mol,均小于-5.0 kcal/mol,表明GLD与MUC2单体和四聚体均具有较好的亲和力[30]。氢键、范德华力、π-烷基(疏水作用的一种)是两分子间的主要作用力。例如:GLD与MUC2-1的CYS-1338等3 个氨基酸残基形成氢键,与ASP-1358等5 个氨酸酸残基通过范德华力产生相互作用。上述结果表明:GLD与MUC2分子间存在较强的相互作用,这可能是造成GLD即使溶解在DMSO中,渗透性仍很差的一个重要原因。在GLD/HP-β-CD包合物中,GLD整个分子处于HP-β-CD的疏水空腔中,无法与MUC2产生相互作用;另一方面,由于HP-β-CD表面亲水,推测其不能与MUC2发生较强的相互作用,因此,GLD/HPβ-CD包合物可以顺利透过肠黏液层。
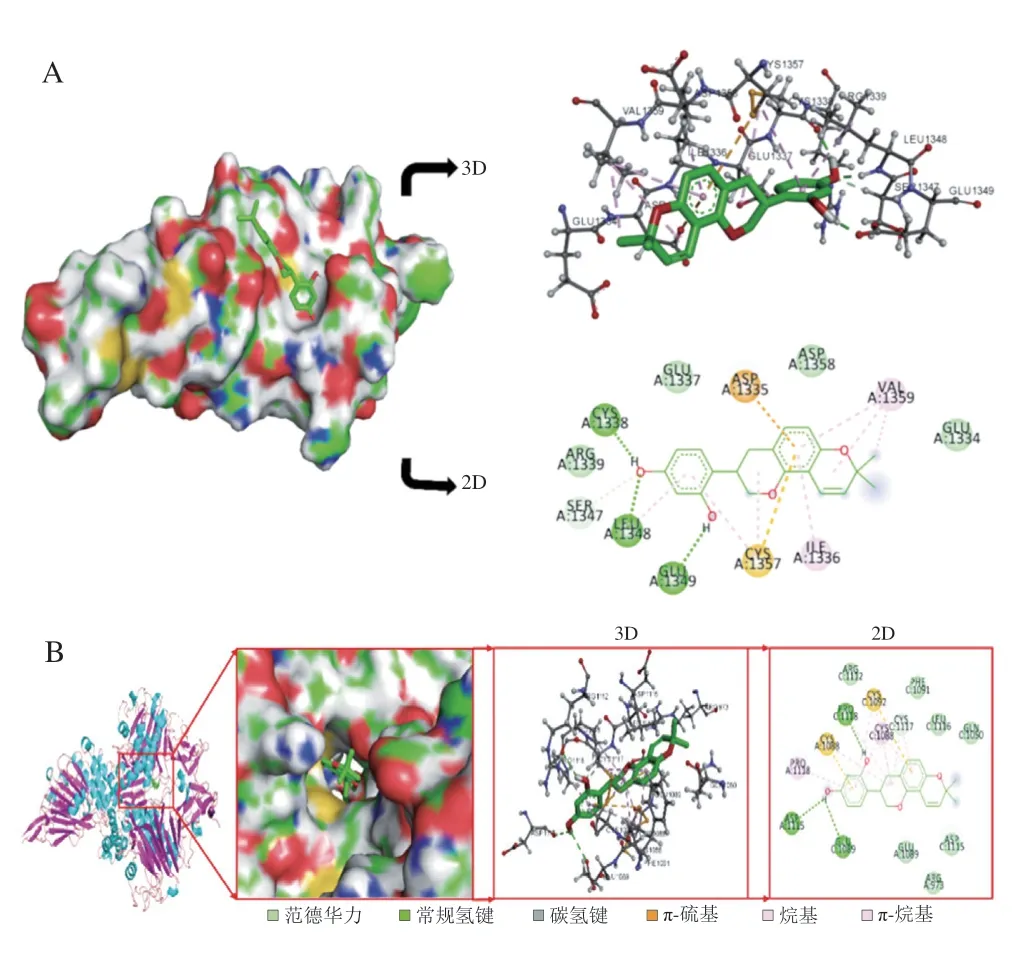
图9 GLD与MUC2分子对接图Fig.9 Molecular docking diagram of GLD and MUC2
2.2.4 Caco-2细胞对GLD的摄取
药物或活性分子被小肠细胞摄取是其吸收过程的又一重要环节;高摄取量通常是高吸收率、高生物利用度的前提和保障。载体可能因改变细胞膜渗透性而影响药物的细胞摄取,进而影响吸收。Caco-2细胞在结构和生化特性上都与小肠上皮细胞相似,因此被国内外广泛地用于研究药物的小肠摄取和吸收[31]。本实验采用Caco-2细胞模拟小肠上皮细胞,研究了GLD/HP-β-CD包合物对GLD细胞摄取的影响。
2.2.4.1 细胞毒性
不同浓度GLD样品处理Caco-2细胞4 h,细胞存活率如图10所示。在10~50 μmol/L范围内,GLD/H2O、GLD/DMSO及GLD/HP-β-CD处理的细胞存活率均在85%以上,基本对细胞无毒副作用[32]。因此,选择浓度50 μmol/L做3 个样品后续的摄取实验。
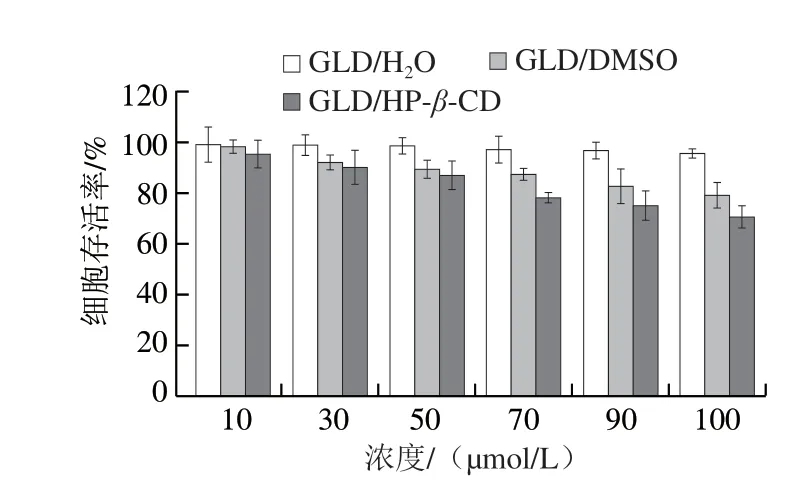
图10 不同浓度GLD样品处理的Caco-2细胞毒性Fig.10 Cytotoxicity of different concentrations of free and encapsulated GLD to Caco-2 cells
2.2.4.2 Caco-2细胞对GLD的摄取量
50 μmol/L的GLD/H2O、GLD/DMSO及GLD/HP-β-CD孵育Caco-2细胞4 h后,Caco-2细胞对样品中GLD的摄取量如图11所示。Caco-2细胞对GLD/H2O组的摄取量最小,为0.039 mg/g;对GLD/DMSO样品的摄取量显著高于GLD/H2O组,约为0.113 mg/g,是GLD/H2O组的2.9 倍。GLD/HP-β-CD的细胞摄取率达0.349 mg/g,比GLD/H2O组提高了795%,比GLD/DMSO组提高了209%。
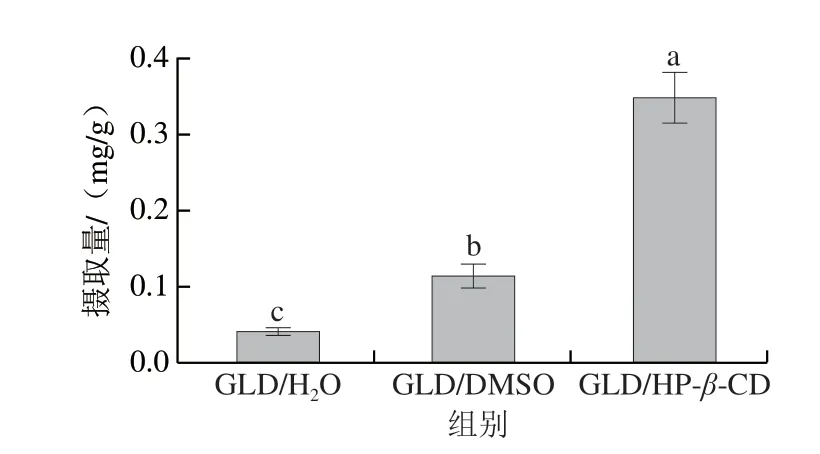
图11 GLD-样品在4 h对Caco-2细胞的摄取量Fig.11 Uptake of free and encapsulated GLD by Caco-2 cells at 4 h
对于GLD/H2O和GLD/DMSO样品,GLD主要以被动扩散的方式通过细胞膜从而被Caco-2细胞摄取。被动扩散方式下,GLD在细胞膜两侧的浓度差是推动其进入细胞的主要动力。DMSO提高了GLD的水溶性,因此Caco-2细胞对GLD/DMSO样品的摄取量远高于GLD/H2O样品。与GLD/H2O相比,GLD/HP-β-CD可以显著提高GLD在细胞培养基中的浓度,因此也可显著提高被动运输方式下小肠上皮细胞对GLD的摄取量。而Caco-2细胞对GLD/HP-β-CD中GLD的摄取量甚至显著高于GLD/DMSO样品,表明除被动运输外,还存在其他的摄取方式,如通过内吞作用进入细胞从而提高细胞的摄取量。
3 结论
制备GLD/HP-β-CD固体包合物,GLD/HP-β-CD中GLD的包合率和载药率分别为90.03%、14.51%,载体HP-β-CD能使GLD在水中的饱和溶解度显著提高至109.36 mg/mL。SEM结果显示GLD/HP-β-CD固体包合物呈形状不规则的片状。DSC结果显示GLD/HP-β-CD包合物中GLD以无定形非晶体结构存在;FTIR及DSC充分证明了HP-β-CD已将GLD包合在空腔内形成了包合物。分子对接显示GLD与HP-β-CD之间的最佳结合能为-7.37 kcal/mol,GLD分子能够完整地进入到HP-β-CD的空腔内,GLD与HP-β-CD分子间形成1 个氢键,GLD与HP-β-CD分子间的相互作用主要靠范德华力维持。GLD/HP-β-CD显著改善了GLD在模拟胃、肠液的溶出速率和累积溶出率,显著提高了在模拟胃、肠液中游离的GLD浓度,极大程度地提高了GLD透过肠上皮表面黏液层的能力,并显著提高小肠上皮细胞对GLD的摄取量。以上研究结果表明:GLD/HP-β-CD具有提高GLD吸收、改善GLD生物利用度的潜力。
