基于机器学习的香豆素衍生物的建模研究
2024-01-03张培俭夏润泽
张培俭, 夏润泽, 高 湛, 刘 壮
(青岛大学计算机科学技术学院, 山东 青岛 266071)
乳腺癌是女性最常见的恶性肿瘤之一,也是女性癌症死亡的主要原因,其发病率随着年龄的增长急剧上升[1-2],全球癌症统计发现,2018年乳腺癌新发病例约208.9万例[3],其中包含死亡病例627 000例,约占全球女性癌症死亡总数的15%[4]。自上世纪60年代,科学家研制出选择性雌激素调制器(SERM),首次被批准用于辅助治疗乳腺癌的SERM是他莫昔芬,该药物已为许多患者提供治疗。虽然SERM改善了ERα阳性乳腺癌患者的症状,但其价格昂贵,且伴有诸多副作用,严重限制了其治疗效果[5],目前活性高但副作用低的SERM尚未发现。近几年,香豆素发展成一种多功能的分子支架,广泛分布在自然界,具有多种药理及治疗作用,如抗细菌,抗真菌,抗疟疾和抗癌等[6]。研究表明,某些香豆素系的杂交体会抑制MCF-7乳腺癌细胞(ER阳性(ER +)人乳腺癌细胞系)的增殖[7],其衍生物对治疗乳腺癌具有重大意义。香豆素衍生物通过对乳腺癌细胞系(MCF-7)半抑制浓度(IC50)值来判断对癌细胞的抑制效果,传统的化学测定方法耗费大量的时间和资源,利用定量构效关系(quantitative structure-activity relationship,QSAR)可以快速准确的预测化合物的IC50。QSAR模型是基于分子结构,通过数学方法预测未经测试的化合物的活性[8-9],GAO Z等[10]通过使用随机森林验证描述符可靠性,并利用混合核函数的支持向量机回归对[1,2,3]三唑[4,5-d]嘧啶衍生物进行抗增殖预测,取得了较好效果;张克俊等人[11]利用基因表达是编程方法建立模型,有效地预测醛类化合物毒性;WANG Y等人[12]使用机器学习方法研究他克林衍生物对乙酰胆碱酯酶抑制剂的活性,为抗阿尔兹海默症药物研发提供了帮助;司宏宗等人[13]利用支持向量机建立预测模型,预测分子结构对药物与血浆蛋白的结合率的影响,对药物筛选提供参考;宋富成等人[14]通过基因表达式编程方法研究离子液体理化结构与青海弧菌Q67毒性关系,取得良好的可靠性。基于此,本文基于启发式算法筛选出的描述符,利用支持向量回归(support vector regression,SVR),广义回归神经网络(general regression neural network,GRNN)和K近邻(K-nearest neighbor,KNN)建立了3种QSAR模型,拟合57种香豆素衍生物对MCF-7乳腺癌细胞的抑制作用并对其作出预测。实验结果表明,利用SVR建立的模型具有较好的预测能力,且模型稳定性最强。该研究为研究治疗乳腺癌的潜在药物分子提供指导。
1 数据
文献[4]、[13]及[15-18]均使用相同的化合物活性IC50测量方法, 即(3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide(简称MTT)方法测量化合物的活性值,获取了57个香豆素衍生物的IC50值,其结构和对应的lg(IC50) 测量值和预测值如表1所示。表中化合物按照约3∶1的比例随机划分为训练集和测试集,lg(IC50)值作为预测标签,标注*符号的为测试集。
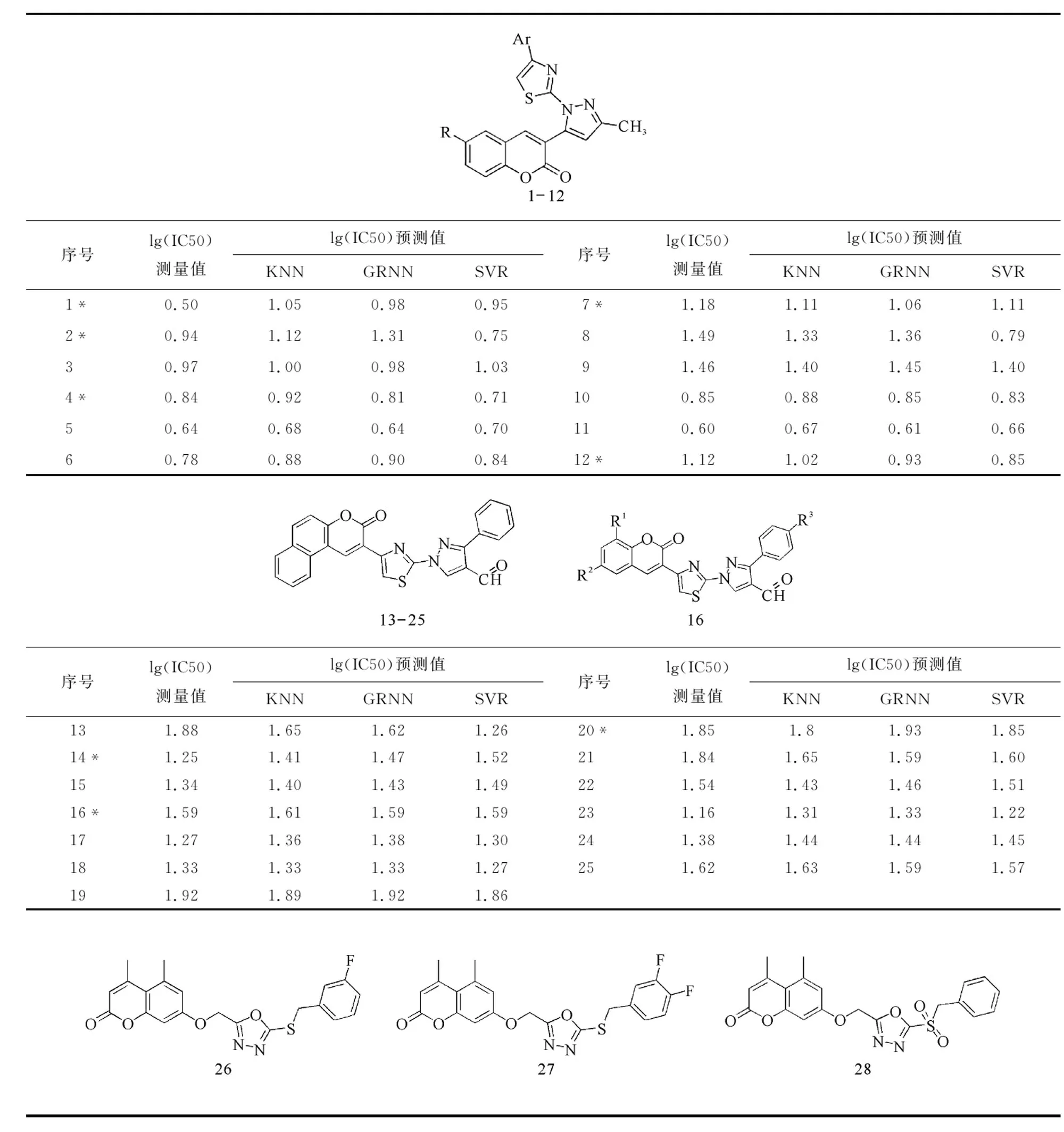
表1 香豆素衍生物及其lg(IC50)测量值和预测值
2 研究方法
2.1 分子描述符
分子描述符的计算步骤如下:
1) 利用ChemDraw软件绘制57个香豆素衍生物的分子结构。
2) 在HyperChem中将分子结构利用MM+分子力学力场进行初步优化,通过半经验AM1和PM3方法进一步优化,获得稳定的分子结构[19]。
3) 将HyperChem中得到的zmt文件导入MOPAC,计算出mno文件[20]。
4) 通过CODESSA程序计算出5类描述符,即构成型、拓扑型、几何型、静电型和量子化学型[21-22]。
利用启发式算法计算描述符,效率高且不受数据集大小的限制,并可自动从描述符空间中选择特征最显著的描述符组,其遵循的规则为:
1) 选择化合物共有的描述符;
2) 排除所有化合物中数值变化很小的描述符;
3) 排除共线描述符,即相关系数大于0.8的描述符。
经计算,本研究最终确定使用3个描述符。
2.2 广义回归神经网络
GRNN是F.S.Donaid在1991年提出的径向基人工神经网络的一种变体,主要用于预测和控制工厂过程建模或一般映射问题[23-24]。GRNN是一种具有高度并行结构的单向学习算法,使多维测量空间中的数据稀疏,也能提供从一个观测到另一个观测的平稳过渡。GRNN结构如图1所示。
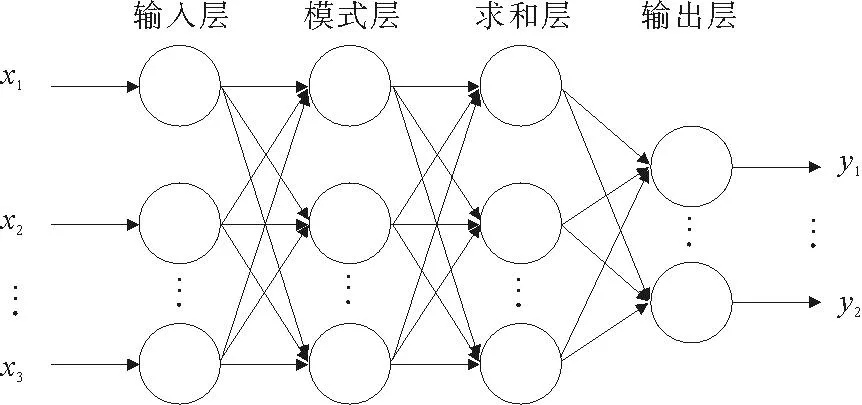
图1 GRNN结构
GRNN由输入层、模式层、求和层和输出层组成[25]。输入层是接收输入信号,神经元数目等于模型的独立特征数量;模式层对输入数据和训练数据集进行必要的映射,模式层中神经元的数量通常等于训练集中的样本数量。模式层节点的输出乘以适当的权重,然后在求和层将所有权重加在一起;输出层节点负责在输入数据集上提供所需的结果。由于预测值(香豆素衍生物IC50的lg值)的维度是1,所以求和层仅包含2个单元,且每个单元连接模式层的所有节点。第1个求和节点将模式层的所有输出求和,并计算式(1)的分子。模式层中的第i个节点与第1个求和节点之间的连接权重等于yi,与第2个求和节点的权重等于1,输出单元计算求和层的2个输出的商,得出系统模型的期望输出值。如果作为输入向量,GRNN的输出值为
(1)
(2)
式中,n是训练数据的数量;x为输入数据,y是期望输出;σ是平滑因子,σ越大,函数近似越平滑,它的取值由具体情况确定[26]。
2.3 K近邻
1991年,Dasarathy[27]提出KNN算法,该算法用于处理多维数据集分类或回归监督学习,根据其最邻近点估算未知点或缺失点的值。最近邻通常被确定为与相邻的未知点的距离最短的点,采用欧式距离度量2个样本之间的距离。在高维数据中,为邻近的变量分配不同的权重是合适的,所以本研究采用高斯函数加权估计的方法计算预测值。欧式距离函数为
(3)
式中,n为数据的特征维度。
2.4 支持向量回归
SVR是支持向量机(support vector machine,SVM)的扩展,SVM成功应用于许多领域的回归分析[34],它是将输入向量映射到高维特征空间,然后在高维特征空间中进行线性回归,利用核函数实现内积运算,降低高维空间中运算的复杂性。
在传统的回归问题中,给定训练集D={(x1,y1),(x2,y2),…,(xm,ym)},其中xi∈R是输入向量,yi∈R是对应的输出值,预测值f(x)与真实值y的差值完全包含在预测结果的损失中。与此不同的是,SVR引入了ε不敏感损失函数,允许在f(x)和y之间最大化ε,提高模型的泛化能力,其中ε表示模型允许的预测误差。因此,SVR的目标函数可表示为
(4)
上式中,ω为超平面系数,C为正则常数,lε为ε不敏感损失函数,其式为
(5)
(6)
在此基础上引入拉格朗日乘子和核函数后,最终目标表达式为
(7)
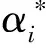
2.5 评价指标
为了验证GRNN、KNN和SVR构建回归模型稳定性的定量指标,R2和均方根误差ERMS的表达式为
(8)
(9)
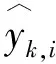
3 结果
根据启发式算法得到3个描述符特征MTICN(min total interaction for a C-N bond)、MBOC(max bond order of a C atom)及NOCl(number of Cl atoms),并由其建立模型,得到MTICN与MBOC对IC50产生正向影响,NOCl则产生负向影响,MBOC系数绝对值为7.852,较另外两者大,故其对IC50具有更大的影响。其公式为
lg(IC50)=0.195d1+7.852d2-0.073d3-16.530
(10)
式中,d1、d2和d3表示3种描述符对应的数据。
3个描述符的物理-化学含义如表2所示,3个描述符的皮尔逊相关系数矩阵如表3所示。

表2 3个描述符的物理-化学含义

表3 描述符相关系数矩阵
本文随机划分数据并建立3个QSAR模型,采用四折交叉验证模型的鲁棒性[39]。
3.1 广义回归神经网络模型
利用GRNN建立QSAR模型,采用高斯函数作为模式层的传递函数,由于高斯函数参数σ的取值影响GRNN的精度,所以必须选取合适的值。为了优化参数,尝试从0.1到1均匀递增变化,最终确定σ为0.1。GRNN训练集和测试集的R2分别为0.949和0.911,RMSE分别为0.092和0.121,GRNN预测结果如图2所示。
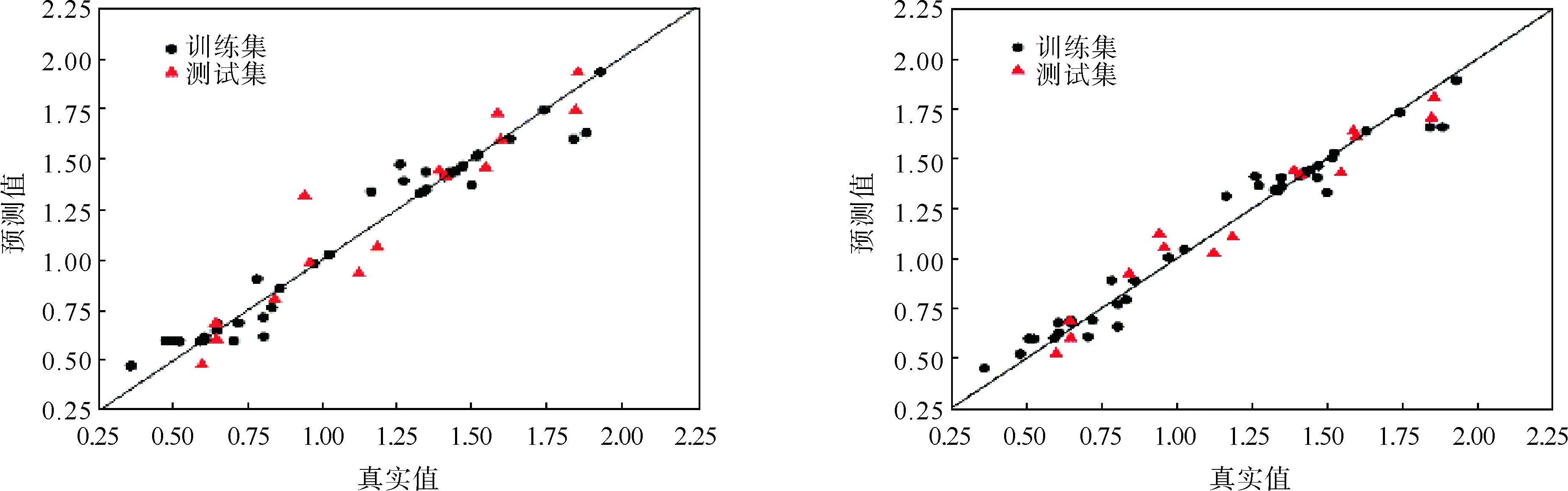
图2 GRNN预测结果
由图2可以看出,利用GRNN构建的模型具有良好的预测能力,其四折交叉验证的训练集和测试集的R2分别为0.957和0.766,RMSE分别为0.083和0.187,表现出过拟合,模型鲁棒性较差。
3.2 K近邻模型
本研究中K取值为3,距离函数为高斯函数,KNN模型预测结果如图3所示。
由图3可以看出,训练集和测试集的R2分别为0.963和0.950,RMSE分别为0.077和0.088,KNN构建的模型也具有良好的预测能力,其四折交叉验证的训练集和测试集的R2分别为0.969和0.794,RMSE分别为0.069和0.185,在交叉验证时,模型表现出过拟合现象,鲁棒性较差。
3.3 支持向量回归模型
SVR建立的模型包括惩罚系数C、ɛ 和 ɛ-不敏感函数、核函数 κ 以及 κ 的相应参数。C为误差的容忍度,大小取决于数据的噪声,而噪声通常是未知的ɛ-不敏感函数允许存在稀疏解,训练后ɛ的最优值为0.1;核函数选用高斯核,形式为F(u,v)=exp(-γ|u-v|2),其中γ为常数,决定了映射到高维空间的特征向量的数量,对训练模型的速度有显著影响。γ越小,支持向量越多,反之亦然,最后采用网格搜索法确定最佳的C和γ:C=1.43,γ=499.768。SVR预测结果如图4所示。
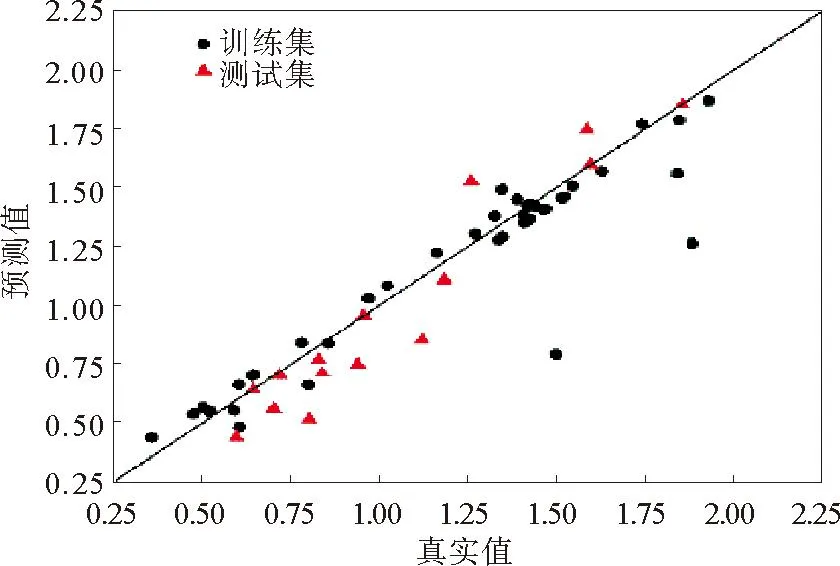
图4 SVR预测结果
由图4可以看出,训练集和测试集的R2分别为0.861和0.829,RMSE分别为0.023和0.068。四折交叉验证训练集和测试集的R2分别为0.886和0.801,RMSE分别为0.142和0.184。因此,SVR构建的模型具有较好的预测能力和最强的鲁棒性,与真实数据较吻合。
3.4 结果分析对比
通过交叉验证对比,SVR模型预测效果良好,具有最好的鲁棒性。KNN模型和GRNN模型虽然表现出很好的预测能力,但是在交叉验证时也表现出了过拟合的趋势,这是由于本研究数据量较小,且特征维度较低导致。
4 结束语
本文通过收集已知化合物结构性质信息建立QSAR模型,更准确地预测未知化合物结构的活性和毒性。在启发式方法下,通过支持向量机、广义回归神经网络和k近邻方法建立3种QSAR模型,使用57种香豆素衍生物对MCF-7细胞的抑制作用进行预测,3种模型预测结果均与实际值吻合较好,且SVR模型最具鲁棒性,表明本文构建的模型对未知乳腺癌药物研发能够提供可靠的活性预测支持。此外,本文采用的MTICN、MBOC和NOCl 3个关键分子描述符对香豆素衍生物活性具有重要影响,为该类衍生物的研究提供方向指引,降低药物研发成本。然而,由于数据集数量限制及机器学习方法缺陷,无法将结论泛化到更多化合物上使用,并难以对香豆素衍生物活性变化进行合理解释,后续将围绕此问题进行进一步研究,为药物筛选提供更多理论指导。
