卡巴匹林钙水分测定法的改进
2024-01-02于晓辉马秋冉
于晓辉,王 雷,杨 星,马秋冉
(中国兽医药品监察所,北京 100081)
卡巴匹林钙收载于《中国兽药典》2020年版一部[1],其中的【水分】项测定方法为:取本品1.0 g,加无水甲醇与二甲基甲酰胺各15 mL,照水分测定法(附录0832第一法A)测定。按照该方法进行测定时发现,结果平行性差、无法准确评价产品中的水分含量,具体实验结果见表1。

表1 现行标准方法的测定结果
从表1中的结果得知,采用现行质量标准中的水分测定方法,对同一批卡巴匹林钙样品进行测定时,数据的平行性、重复性很差。第一次测定结果中的最大值和最小值的相对偏差高达82%;第二次测定结果中,第三份样品的仪器测定结果为0.00%,但在实际测定过程中明确观察到样品消耗了滴定液,出现0.00%的结果,表明水分测定仪的终点判断出现了误差。
水分是卡巴匹林钙质量控制的重要项目,当其中的水分值超过限度0.2%时,即会造成卡巴匹林钙加速降解,生成水杨酸等一系列降解杂质,严重影响产品的质量[2,3]。现行质量标准中的方法无法真实反映卡巴匹林钙中的水分情况,无法保证卡巴匹林钙质量可控。
为了更好的控制卡巴匹林钙的质量,我们对现行质量标准中的水分测定法进行了改进。
1 材料与方法
1.1 仪器与试剂 701KF卡氏水分滴定仪,瑞士万通公司;费休氏试液(滴定度1 mg/mL),Honeywell公司;二甲基甲酰胺,Alfa Aesar公司;无水甲醇:Honeywell公司。
1.2 试药 供试品:共4家企业生产的4批卡巴匹林钙。
1.3 实验方法 向滴定杯中分别加入二甲基甲酰胺20 mL、无水甲醇20 mL,取供试品0.1 g,精密称定,置于滴定杯中,搅拌5分钟,照水分测定法(附录0832第一法A)测定。注:连续测定4份样品后,需更换滴定杯中的溶剂。
2 结果与分析
2.1 溶剂中二甲基甲酰胺与无水甲醇比例的筛选通过前期的实验研究得知,二甲基甲酰胺-无水甲醇适合作为卡巴匹林钙水分测定的溶剂。其中,二甲基甲酰胺作为分散剂,起到分散样品、“释放”水分的作用,无水甲醇参与滴定反应,起到促进反应顺利进行的作用。二者“各司其职”,在一定比例范围内协同作用,才能得到准确可靠的测定结果。
因此,本研究对二甲基甲酰胺和无水甲醇的比例进行了比较研究,按照1.3中的方法,考察在不同比例的溶剂中,分别搅拌5 min和搅拌10 min测定结果的异同。为保证研究充分、结果可靠,我们将上述过程重复测定,即,每种混合溶剂中,连续测定样品4份,第一份搅拌5 min、第二份搅拌10 min、第三份搅拌5 min、第四份搅拌10 min。实验结果见表2。
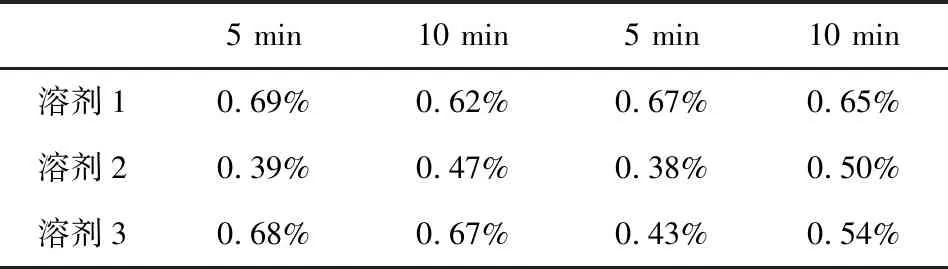
表2 二甲基甲酰胺-甲醇不同比例时的测定结果
溶剂1:二甲基甲酰胺20 mL、无水甲醇20 mL(1∶1)
溶剂2:二甲基甲酰胺13 mL、无水甲醇27 mL(1∶2)
溶剂3:二甲基甲酰胺27 mL、无水甲醇13 mL(2∶1)
从表2的实验结果得知,当以溶剂2为溶剂时,搅拌5 min后的测定结果与搅拌10 min后的测定结果存在差异,搅拌10 min后的测定结果高于搅拌5 min后的。这可能是由于该混合溶剂中,二甲基甲酰胺的量较少、分散能力较弱,搅拌5 min不足以充分“释放”卡巴匹林钙中的水分。
当以溶剂3为溶剂时,前两份样品,即分别搅拌5 min和10 min的测定结果一致;但此后,搅拌5 min和搅拌10 min的测定结果出现差异,并且第三、第四份样品的测定结果低于第一、第二份样品的结果。这可能是由于溶剂3中,甲醇量较少,随着样品量的增加,参与滴定反应的溶剂量减少,滴定反应无法充分彻底进行,以至于测定两份样品后,后续样品中的水分无法被充分滴定,测定结果偏低。
当以溶剂1,即二甲基甲酰胺-甲醇(1∶1)为溶剂时,实验结果稳定,平行性良好,搅拌5 min和搅拌10 min的测定结果基本一致,重复测定4份样品的结果也一致。
上述实验结果表明,改进后的方法中,二甲基甲酰胺和无水甲醇的比例应为1∶1。
2.2 加入样品后搅拌时间的选择 卡巴匹林钙在二甲基甲酰胺-无水甲醇中的溶解性较差,为保证其在上述溶剂中被充分分散、样品中的水分被充分“释放”出来,加入样品后、开始滴定前应对溶剂杯中的样品充分搅拌。
本研究按照1.3中的方法,对样品加入滴定杯后的搅拌时间进行了比较研究,结果见表3。

表3 不同搅拌时间的测定结果
从表3的实验结果得知,加入样品后立即滴定(0 min)的水分值明显低于搅拌一定时间后再滴定的结果;搅拌5 min后再滴定的结果与搅拌10 min、15 min、20 min的结果一致,实验研究结果表明,搅拌5 min即能将卡巴匹林钙样品中的水分充分“释放”出来。所以,本研究最终选择加入样品后搅拌5 min再进行滴定。
2.3 取样量的选择 经过前期实验研究得知,将取样量降低至0.1 g并以二甲基甲酰胺+无水甲醇为溶剂时,测定结果稳定、平行性良好。为了更好的确定卡巴匹林钙的取样量,我们按照1.3中的方法,比较研究了0.05 g、0.1 g和0.2 g三种取样量,结果见表4。
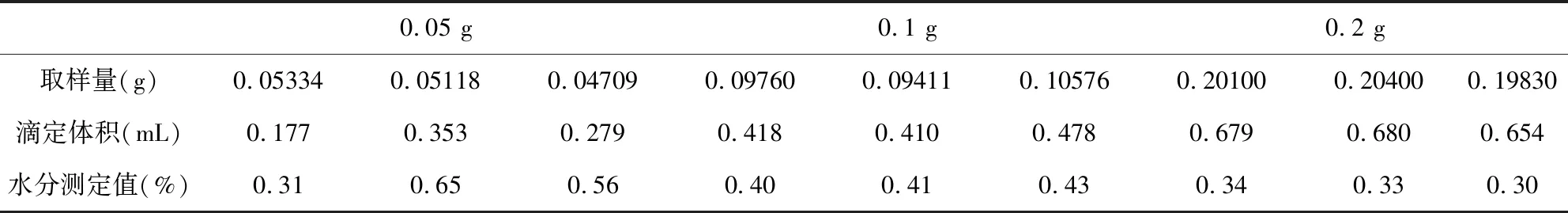
表4 不同取样量时的测定结果
从表4的实验结果得知,当取样量为0.05 g时,数据的平行性较差,最大值和最小值的相对偏差约为35%。所以,0.05 g不适合作为该方法的取样量。
当取样量为0.2 g时,数据的平行性良好,最大值与最小值的相对偏差约为6%。但是,0.2 g取样量时,一份样品测定完成后,仪器约需5 min才能达到平衡状态(condition),这可能是由于取样量较大,样品中的水分未能在短时间内被完全滴定,从而造成滴定延迟。
当取样量为0.1 g时,测定结果稳定,数据平行性良好,最大值与最小值的相对偏差约为4%;一份样品测定完成后,仅需约1 min,仪器即达到平衡状态(condition)。所以,本研究最终选择0.1 g作为改进方法的取样量。
2.4 连续测定样品份数验证 通过前期实验发现,随着滴定杯中样品量的逐渐增加,连续测定多份样品时,结果的平行性和准确性有变差的趋势。为保证连续测定时结果准确,我们按照1.3中的方法,对连续测定样品的份数进行了考察,实验结果见表5。

表5 连续测定6份样品时的测定结果
从表5的实验结果得知,在40 mL溶剂中,连续测定6份样品时,前5份结果的平行性良好,最大值和最小值的相对偏差约为5%;第6份结果的准确性出现变差趋势,这可能是由于加入的样品量较大,超出了40 mL溶剂的分散能力和反应能力,此时需要更换滴定杯中的溶液,重新加入溶剂后再进行测定。
为保证连续测定结果的可靠性,改进后的方法规定“连续测定4份样品后,需更换滴定杯中的溶剂。”
2.5 滴定杯中溶剂量的选择 通过前期实验得知,溶剂量越大,可以测定的样品份数越多、越容易得到平行性良好的实验结果。
常见滴定杯的容量约为100 mL,如果向其中加入的溶剂量超过50 mL,随着费休氏试液标定和样品测定实验的进行,容易出现杯中溶剂量过多、杯中溶液倒吸等现象。
综合考虑,本研究最终选择40 mL溶剂量进行实验。
2.6 样品测定 按照1.3中的方法对四批样品进行了测定,结果见表6。
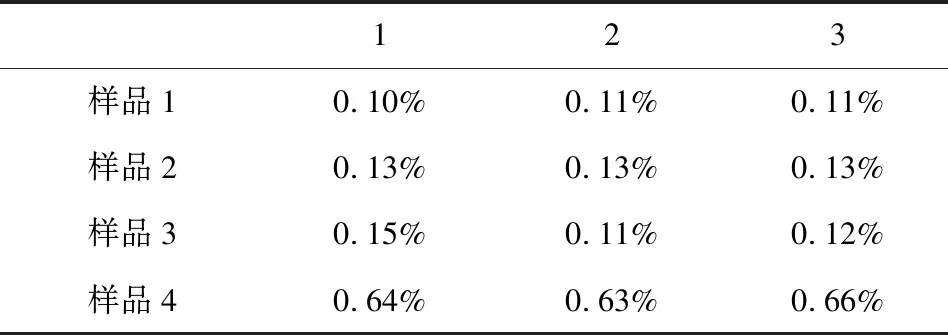
表6 应用改进后方法的测定结果
从表6的实验结果得知,应用改进后的水分测定法对四批样品进行测定,结果稳定,数据平行性良好,表明改进后的方法彻底解决了现行质量标准中卡巴匹林钙水分测定法存在的数据平行性差、结果混乱异常的问题。
2.7 与其他水分测定法的比较 为了进一步验证改进后方法的准确性,我们分别采用60 ℃减压干燥失重法和气相色谱法(《中国兽药典》附录0832 第三法)对四批卡巴匹林钙样品中的水分进行测定。结果见表7和表8。
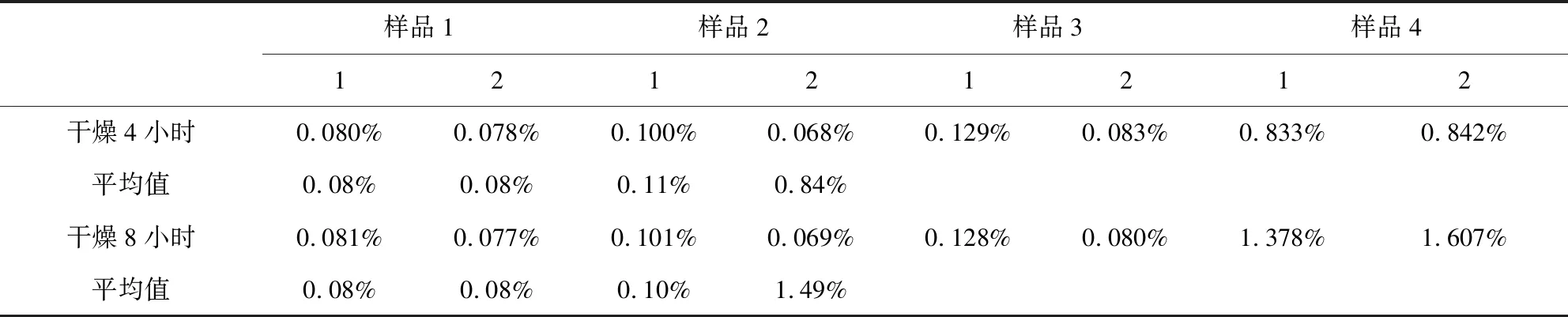
表7 减压干燥失重法水分测定结果
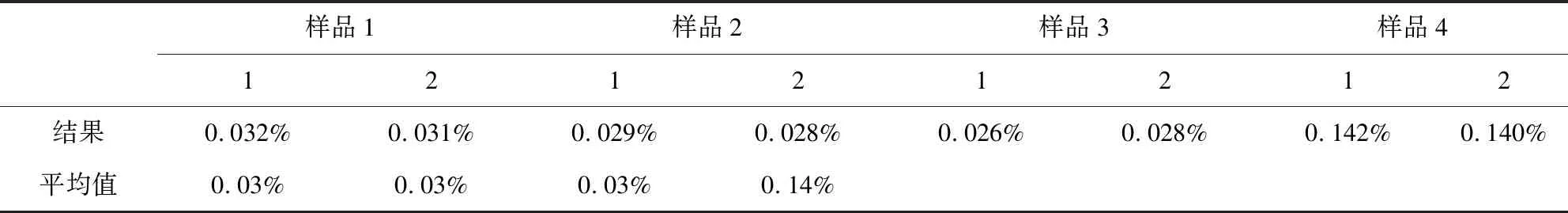
表8 气相色谱法水分测定结果
卡巴匹林钙对温度敏感,在高温下易产生降解,生产企业通常采用80 ℃的温度对生产得到的原料进行干燥。为保证测定结果准确,本研究采用60 ℃减压干燥的方式对4批卡巴匹林钙样品进行干燥失重测定。
从表7的实验结果得知,采用60 ℃减压干燥失重法测定时,前三批产品的测定结果与改进后方法的测定结果一致,均为0.1%;样品4的测定结果(0.8%~1.5%)与改进后方法的测定结果(0.6%)差异明显。
之所以出现上述差异,是由于被测样品的降解程度不同造成的。通过HPLC实验结果[1,4]得知,前三批样品中杂质含量很少,主要杂质-水杨酸的含量约为0.2%~0.4%;而样品4中杂质含量多,主要杂质-水杨酸的含量约为20%。水杨酸在70 ℃的条件下即会发生升华,在60 ℃减压干燥条件下,样品4中的水杨酸不断升华,从而造成其干燥失重结果高于改进后方法的测定结果,并且其干燥8小时后仍未达到恒重。前三批样品测定过程中,受到杂质干扰少,所以,这三批样品的干燥失重结果与改进后方法的测定结果一致。
上述实验结果表明,当卡巴匹林钙样品合格、杂质水杨酸的量较少时,60 ℃减压干燥的方式可以准确测定样品中的水分值。但当样品降解严重、杂质水杨酸的量较多时,60 ℃减压干燥的方法不能准确测定其中的水分。
所以,60 ℃减压干燥失重法用作卡巴匹林钙的水分测定时,适用范围较窄。
从表8的实验结果得知,采用气相色谱法测得的卡巴匹林钙样品中的水分值明显低于改进后方法的测定结果,这是由于热导检测器的灵敏度较低造成的。所以,附录0832第三法 气相色谱法不适合作为水分限度值较低的卡巴匹林钙水分测定方法。
3 讨 论
为了保证实验结果真实可靠,我们进行了如下三方面的工作:为了排除样品不均匀对测定结果造成影响,实验开始前,我们将样品充分混匀;为了排除溶剂“不新鲜”而对测定结果产生影响,每次实验开始前,我们均更换滴定杯中的溶剂;为了排除仪器不稳定对测定结果造成影响,每次测定样品前,我们均应用该仪器对费休氏试液进行标定,当连续三次标定结果相对偏差≤1.0%时,才开始样品测定。
卡尔费休氏滴定反应受pH值影响,5~7.5是理想的pH值范围。我们曾以为卡巴匹林钙偏酸性是造成测定结果平行性差的原因,但是,当我们向溶剂中添加了20%~30%的咪唑缓冲盐后,实验结果仍然不理想,最大值和最小值的相对偏差在20%~30%的范围内。验证结果表明,卡巴匹林钙偏酸性不是现行标准方法测定结果平行性差的主要原因。
通过实验验证得知,现行标准方法测定结果平行性差的主要原因是卡巴匹林钙中的Ca2+与费休氏试液滴定过程中产生的SO42-结合形成CaSO4沉淀, CaSO4沉淀颗粒细小、附着力强,当卡巴匹林钙样品量大(1.0 g)时,生成的CaSO4沉淀量亦较大,滴定杯中的溶剂无法充分分散生成的沉淀,以致沉淀覆盖电极表面,造成电极灵敏度变差、无法正常指示滴定终点,从而导致滴定结果异常。
甲酰胺是费休氏水分测定法中最推荐使用的强分散剂,但是,我们通过实验研究证明,甲酰胺不适于用作卡巴匹林钙水分测定时的分散剂。
采用水分测定仪进行测定时,当滴定体积占滴定管总体积的10%~90%,即可保证滴定结果的准确性。目前,水分测定仪滴定管的最小体积为1 mL,所以,当滴定体积大于0.1 mL即可满足方法准确性的要求。根据实验结果推算得知,当水分测定值为0.2%时,0.1 g取样量消耗的滴定液(滴定度1 mg/mL)体积约为0.2 mL,满足方法准确性的要求。
溶剂二甲基甲酰胺、无水甲醇应于实验开始前分别加入滴定杯中,不可将二者提前混合在一起,因为甲酰胺类化合物易与醇类化合物发生副反应,尤其在光照条件下,二者的副反应会加速发生,以致滴定杯中的溶液呈现“果冻状”,严重影响实验的顺利进行。
虽然溶剂中采用二甲基甲酰胺作为分散剂,以减少滴定反应过程中生成的CaSO4沉淀对电极的覆盖,但是,为了更好的去除电极表面可能覆盖的沉淀,建议实验结束后,用粗糙的滤纸等擦拭电极铂片,以保持电极的灵敏度。
本研究未采用卡式炉升温的方式提取样品中的水分。因为卡巴匹林钙高温下易降解,主要降解产物是水杨酸,水杨酸在70 ℃发生升华。如果采用卡式炉升温加热的方式对卡巴匹林钙中的水分进行提取,会造成降解产物水杨酸进入水分测定系统中,从而对水分测定结果产生明显干扰。
本研究采用容量法对卡巴匹林钙中的水分进行测定,而未采用库伦法,主要原因有以下两方面:①经实验验证,容量法的测定结果准确可靠,满足卡巴匹林钙质量控制的要求;②相比于库伦法水分测定仪,容量法水分测定仪造价低廉,普及率广,尤其在各企业实验室中应用广泛。为节约企业生产成本,最终改进后的方法采用了容量法。
采用杂质含量较多的样品4进行实验研究。卡巴匹林钙易降解生成水杨酸,而水杨酸对水分测定结果干扰明显。为保证改进后的方法具有良好的耐用性,本研究采用杂质含量较多、尤其水杨酸含量较多的样品4进行方法开发和方法验证。
4 结 论
研究改进了卡巴匹林钙水分测定法,解决了现行标准方法数据平行性差、无法反映产品中水分的真实情况等问题。
相比于减压干燥失重法和气相色谱法,改进后的方法适用范围广、灵敏度高、易于推广普及,适用于卡巴匹林钙中的水分测定。
