喹诺酮杂合体的抗耐甲氧西林金葡菌活性研究进展
2023-12-26王林硕李晓王宝君
王林硕 李晓 王宝君

摘要:耐甲氧西林金葡菌(MRSA)是引起院内和社区感染的重要病原菌,高发病率与致死率使得其位居感染病原菌之首。MRSA是多重耐药致病菌,目前已对包括万古霉素在内的几乎所有抗生素产生了不同程度的耐药性。因此,开发新型抗MRSA药物刻不容缓。喹诺酮尤其是氟喹诺酮是临床上广泛使用的一类广谱抗菌药,对包括某些MRSA在内的多种致病菌引起的感染具有良好的疗效。杂合体具有多个药效团可同时作用于不同的药物靶点,故杂合体策略是克服耐药性的常用策略。喹诺酮与其它抗MRSA药效团相结合所得的杂合体可发挥协同作用,寻找新型抗MRSA药物的潜力分子。近年来,药物化学家设计合成评价了多个系列喹诺酮杂合体的抗MRSA活性,发现了多个具有进一步研究价值的潜在化合物。本文将着重介绍2015—2022年间所发展的具有抗MRSA活性的喹诺酮杂合体的研究进展,并归纳构-效关系,为进一步合理设计此类杂合体提供一定的理论支持。
关键词:喹诺酮;杂合体;抗菌;耐甲氧西林金葡菌;构-效关系
中图分类号:R978.1 文献标志码:A
Recent progress of quinolone hybrids with antibacterial potential against methicillin-resistant Staphylococcus aureus
Wang Linshuo1, Li Xiao2, and Wang Baojun1
(1 Shijiazhuang Medical College, Shijiazhuang 050599; 2 Handan Maternal and Child Health Care Hospital, Handan 056000)
Abstract Methicillin-resistant Staphylococcus aureus (MRSA) strains, the most common pathogens in hospital and community, remains a leading cause of bacterial infections worldwide due to the high mortality and morbidity. MRSA are multi-drug resistant pathogens and have already developed resistance to almost all antibiotics including vancomycin, necessitating the development of novel agents for combating these superbugs. Quinolones, especially fluoroquinolones, are broad-spectrum antibacterial agents widely applied in clinical practice and demonstrated excellent curative effect on infections caused by diverse pathogenic bacteria, including some MRSA. Hybrid molecules possess multiple pharmacophores that can act on different drug targets simultaneously, so hybridization is a promising strategy to overcome drug resistance. Combination of the quinolone moiety with other pharmacophores against MRSA has the potential to exert synergistic effect, and thus quinolone hybrids are useful templates for searching for new anti-MRSA drugs. In this review, the anti-MRSA potential and the structure-activity relationship of quinolone hybrids are discussed, which will provide a reference for further research.
Key words Quinolone; Hybrid; Antibacterial; MRSA; Structure-activity relationship
耐甲氧西林金葡菌(MRSA)是一種传染性和致病性极强的共生菌,可导致肺炎、菌血症、心内膜炎、皮肤和软组织感染、骨和关节感染甚至死亡,是院内最常见的致病菌[1-2]。MRSA呈多重耐药性,也称“超级细菌”,对所有与甲氧西林相同结构的β-内酰胺类和头孢类抗生素均耐药,对其它抗菌药如氨基糖苷类、大环内酯类、四环素类、氟喹喏酮类、磺胺类和利福平等具有不同程度的耐药[3-4]。目前,MRSA感染几乎遍及全球,并已超过乙肝和艾滋病,被列为世界3大最难解决感染性疾患的首位[5-6]。MRSA感染的治疗是临床最棘手的难题之一,最常用也是疗效最肯定的抗菌药有万古霉素、去甲万古霉素和替考拉宁、利奈唑胺、特地唑胺和康替唑胺等[7-8]。然而,新型耐药MRSA如万古霉素中介金葡菌(VISA)、异质性万古霉素中介金葡菌(h-VISA)和万古霉素耐药金葡菌(VRSA)的出现,使得临床医师可能会在不久的将来面临无药可选的窘境[9-10]。因此,开发新型抗MRSA药物刻不容缓。
喹诺酮尤其是氟喹诺酮具有抗菌谱广、抗菌活性强、口服吸收好、组织浓度比较高和不良反应较低等诸多优点,广泛应用于临床治疗各种致病菌引起的感染[11-12]。其中,某些第四代喹诺酮如莫西沙星、德拉沙星、西他沙星、加雷沙星、奈诺沙星和奥泽沙星等目前已在临床上用于治疗MRSA感染[13-14]。因此,喹诺酮骨架是开发新型抗MRSA药物的潜在结构单元。将两个或多个药效团糅合到一个分子中所得的杂合体可同时作用于细菌的多个靶点,具有改善药动学性质、降低毒副作用、提高药效、拓展抗菌谱和克服耐药性的潜力[15-16]。因此,将喹诺酮与其它抗MRSA药效团相结合所得的杂合体可发挥协同作用,是寻找新型抗MRSA药物的潜力分子[17-18]。
近年来,药物化学家设计、合成了多个系列喹诺酮杂合体,并评价了它们的抗MRSA活性。其中的某些杂合体如氟喹诺酮-利福霉素杂合体Ro-23-9424 (Roche公司,可通过阻扰细菌合成细胞壁和干扰细菌核酸的功能发挥抗菌活性,已经进入Ⅱ期临床试验阶段)和氟喹诺酮-β-内酰胺杂合体CBR-2092 (Cumber公司,可通过抑制RNA的合成和DNA促旋酶及拓扑异构酶IV发挥抗菌活性,已经进入Ⅱ期临床试验阶段)显示出良好的体内外抗MRSA活性,具有进一步研究价值。本文将着重介绍2015—2022年间所发展的具有抗MRSA活性的喹诺酮杂合体的研究进展,并归纳构-效关系,为进一步合理设计此类杂合体提供一定的理论支持.
1 喹诺酮杂合体的抗MRSA活性
氟喹诺酮-嘧啶杂合体1(图1;最小抑菌浓度/MIC: 1.0~>256 μg/mL)的抗MRSA构-效关系研究表明:①氟喹诺酮N-1位取代基对活性有显著影响,且乙基优于环丙基;②向嘧啶的C-4位引入供电子的氨基对活性有利,而吸电子的三氟甲基对活性不利[19]。其中,杂合体1a~c(MIC: 1.0 μg/mL)的抗MRSA活性是环丙沙星(MIC: 2.0 μg/mL)和诺氟沙星(MIC: 8.0 μg/mL)的2和8倍。值得一提的是,杂合体1a(MIC: 0.5~1.0 μg/mL)对所测3株药敏型金葡菌也具有良好的活性,且活性与环丙沙星(MIC: 0.5~2.0 μg/mL)和诺氟沙星(MIC: 2.0~4.0 μg/mL)相当或更优。此外,杂合体1a(半数细胞毒性浓度/CC50: >
50 μg/mL)对正常HaCat细胞的毒性较低。作用机制研究表明,杂合体1a不仅可通过抑制生物膜的形成快速发挥抗菌活性,而且还可通过提高胞内活性氧浓度和形成DNA-1a复合物促使细菌凋亡。计算机模拟发现,杂合体1a具有良好的吸收、分布、代谢和消除(ADME)性质,且与诺氟沙星和环丙沙星相当。不仅如此,与诺氟沙星和環丙沙星相比,该杂合体产生耐药性的几率更低。
绝大多数喹诺酮-嘧啶杂合体2(MIC: 0.75~>50 μg/mL)和3(MIC: 0.38~12.5 μg/mL)显示出潜在的抗MRSA活性,且构-效关系显示:①向嘧啶C-4位的苯环对位引入小体积烷基尤其是甲基可增强活性;②嘧啶与苯环之间嵌入氧原子对活性有利;③喹诺酮N-1位为环丙基时活性优于相应的直链或支链烷烃衍生物[20]。其中,杂合体2a,b(MIC: 0.75 μg/mL)和3a,b(MIC: 0.38 μg/mL)是各系列中活性最高的化合物,且活性与环丙沙星(MIC: 0.50 μg/mL)相当,而优于万古霉素(MIC: 1.5 μg/mL)。进一步评价发现,杂合体3b(MIC: <
0.17~0.69 μg/mL)对5株临床分离MRSA也显示出良好的活性,且活性高于万古霉素(MIC: 0.78~3.13 μg/mL)。不仅如此,该杂合体(MIC: 0.125~1.0 μg/mL)对耐左氧氟沙星MRSA和耐万古霉素中度耐药金葡菌(VISA)的活性是左氧氟沙星(MIC: 16~>64 μg/mL)和万古霉素(MIC: 2.0~>8.0 μg/mL)的≥8倍。特别值得一提的是,杂合体3b在MRSA传代15次后MIC没有任何增加,而对照药万古霉素则提升了4倍,提示该杂合体不易产生获得耐药性。对接试验表明,该杂合体的氨基嘧啶结构片段可与DNA碱基形成2个氢键,这可能是该杂合体克服耐药性的原因。
喹诺酮-嘧啶二酮杂合体4(MIC: 15.4 μg/mL)及其衍生物5(MIC: 24.2 μg/mL)的抗MRSA AUMC 261活性优于环丙沙星(MIC: 188.6 μg/mL)[21]。作用机制研究发现,杂合体4(IC50:1.72和4.36 μmol/L)和5(MIC: 5.72和7.77 μmol/L)对DNA促旋酶和拓扑异构酶IV具有双重抑制作用。因此,二者可作为抗MRSA先导物进行进一步结构优化。
环丙沙星-喹唑啉酮杂合体6(MIC: 8.0~32 μg/mL)的抗MRSA活性普遍优于沙拉沙星-喹唑啉酮杂合体7(MIC: 16~32 μg/mL),提示氟喹诺酮母核对活性有一定影响[22]。进一步研究发现,向喹唑啉酮的C-6位引入供电子基如甲氧基对活性有利,而吸电子基则对活性不利;向C-7位引入卤素也会降低活性。其中,杂合体6b(MIC: 8.0 μg/mL)的抗MRSA活性是环丙沙星(MIC: 16 μg/mL)的2倍。此外,杂合体6b(MIC: 8.0 μg/mL)的抗药敏型金葡菌活性是环丙沙星(MIC: 32 μg/mL)的4倍,且耐药指数(RI: MICMRSA/MICMSSA)为1.0,提示该杂合体无交叉耐药性。
2-喹诺酮-喹唑啉酮杂合体8对所测5株甲氧西林敏感型金葡菌(MSSA)、3株MRSA和3株VISA的MIC分别为4.0~8.0 μg/mL、4.0~8.0 μg/mL和4.0~16 μg/mL [23]。进一步研究发现,该杂合体(半数抑制浓度/IC50:1.21 μmol/L)具有潜在的抑DNA促旋酶亚基B(gyrB)活性。代谢稳定性研究表明,该杂合体在血浆和肝微粒体的半衰期分别为>372.8 min和24.5 min。总之,杂合体8可作为gyrB抑制剂进行深入研究。
莫西沙星/磺酰胺杂合体9的抗MRSA构-效关系显示:①向苯环的对位引入强吸电子基如硝基对活性不利,而卤素和甲氧基对活性影响不大;②磺酰基并非这类杂合体具有高活性所必需的基团,用羰基或者次甲基取代活性并不会降低[24]。其中,杂合体9a~e(MIC: <2.64 μg/mL)的抗MRSA活性与环丙沙星(MIC: 1.31 μg/mL)处于同一水平,值得进一步研究。
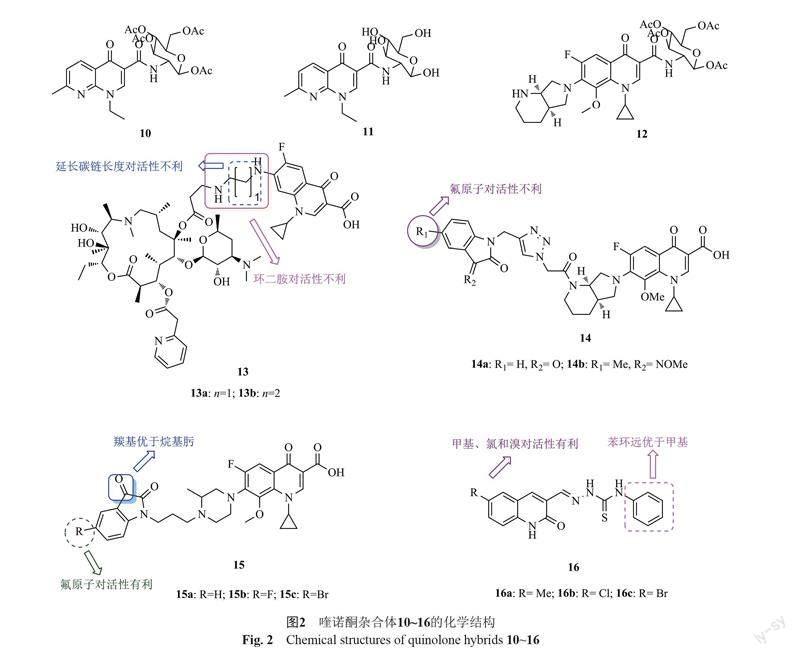
萘啶酮-葡萄糖杂合体10(图2,MIC: 64 μg/mL)和11(MIC: 64 μg/mL)及莫西沙星-葡萄糖雜合体12(MIC: 128 μg/mL)的抗MRSA活性较弱,远逊于对照药莫西沙星(MIC: <0.2 μg/mL)[25-26]。这可能是由于氟喹诺酮的C-3位羧酸结构片段和C-4位羰基结构单元是DNA促旋酶的结合位点,对喹诺酮的抗菌活性至关重要,对该位点进行修饰往往会导致活性的降低甚至消失所致[11]。
氟喹诺酮-阿奇霉素杂合体的抗红霉素核糖体甲基化酶基因(erm)编码诱导耐药的MRSA PU32构-效关系显示:①氟喹诺酮与阿奇霉素母核之间用直链烷基二胺连接优于环二胺;②碳链长度与活性息息相关,且延长碳链长度对活性不利;③将喹诺酮C-7位的氨基连接子换成炔基连接子且移至C-6位将导致活性大幅下降[27]。作用机制研究显示,这类杂合体可通过抑制蛋白质合成和抑制DNA复制双重作用机制发挥抗菌活性。其中,杂合体13a,b(MIC: 8.0 μg/mL)抗MRSA PU32的活性是对照药阿奇霉素(MIC: 32 μg/mL)和环丙沙星(MIC: 64 μg/mL)的4和8倍,值得进一步结构优化。
莫西沙星-1,2,3-三氮唑-靛红杂合体14(MIC: 1.0~32 μg/mL)具有潜在的抗MRSA活性,且构-效关系显示,向靛红的C-5或C-7位引入氟原子或向C-3位引入乙肟对活性不利[28]。代表物14a,b(MIC: 1.0 μg/mL)
的抗MRSA活性可与万古霉素(MIC: 1.0 μg/mL)相媲美,而是母药莫西沙星(MIC: 8.0 μg/mL)的8倍。细胞毒性试验表明,二者(CC50: 64和32 μg/mL)对正常CHO细胞的毒性较低。体内药动学研究表明,杂合体14b(100 mg/kg, 皮下注射)的半衰期为3.1 h,达峰时间为29 min。
加替沙星-靛红杂合体15(MIC: 0.125~0.5 μg/mL)具有优秀的抗MRSA活性,且活性不逊于加替沙星(MIC: 0.25 μg/mL)和环丙沙星(MIC: 0.5 μg/mL) [29]。构-效关系显示,与羰基相比,向靛红的C-3位引入烷基肟对活性不利;向C-5位引入氟原子对活性有利,但溴原子的引入不会增强活性;向加替沙星C-3位的羧酸引入第二个靛红结构片段对活性不利。其中,代表物15b(MIC: 0.125 μg/mL)的抗MRSA活性是加替沙星(MIC: 0.25 μg/mL)和环丙沙星(MIC: 0.5 μg/mL)的2和4倍。
2-喹诺酮-氨基硫脲杂合体的抗MRSA构-效关系显示:①氨基硫脲部分苯环对活性至关重要,将苯环用甲基替代将导致活性大幅降低;②向2-喹诺酮母核的C-6位引入甲基、氯和溴对活性有利,但氟对活性不利[30]。其中,杂合体16a~c(MIC: 312 μg/mL)的活性最高,但弱于环丙沙星(MIC: 62.5 μg/mL)。
2-喹诺酮-噻二唑杂合体17(图3,MIC: 0.25~4.0 μg/mL)对所测的9株临床分离MRSA具有优秀的活性,且构-效关系显示:①2-喹诺酮的N-1位为乙基时杂合体的活性优于相应的甲基和丙基衍生物;②向噻二唑的C-3位引入杂环尤其是噻唑和吡啶可提高活性[31]。
其中,杂合体17a~c(MIC: 0.25~1.0 μg/mL)对所测9株临床分离MRSA的活性普遍优于万古霉素(MIC: 1.0~8.0 μg/mL)。特别值得一提的是,杂合体17a(MIC: 0.25~1.0 μg/mL)对所测9株临床分离MRSA、1株MSSA、2株甲氧西林敏感表皮葡萄球菌(MSSE)、3株耐甲氧西林表皮葡萄球菌(MRSE)和耐万古霉素肠球菌(VRE)具有优秀的广谱活性,且活性是万古霉素(MIC: 1.0~64 μg/mL)的2~128倍。在MRSA感染小鼠模型中,杂合体17a组小鼠的存活率呈计量相关性,且在口服给药9.0 mg/kg时小鼠的存活率为87.5%,而安慰剂组的存活率仅为12.5%。此外,杂合体17a的血浆结合率为99.9%。体内药动学研究表明,杂合体17a(10 mg/kg, 口服给药)的半衰期为2.3 h,达峰时间为1.0 h,药时曲线下面积(AUCinf)为2620 ng·h/mL且最大血药浓度(Cmax)为
827.1 ng/mL。特别值得一提的是,杂合体17a在MRSA传代20次后MIC没有任何增加,提示该杂合体不易产生获得耐药性。作用机制研究发现,DNA促旋酶B是该杂合体的作用靶点。总之,杂合体17a可作为候选物进行深入的临床前研究。
含有肟基的环丙沙星/诺氟沙星-噻唑杂合体(MIC: 0.25~4.0 μg/mL)具有良好的抗MRSA活性,且活性是环丙沙星(MIC: 8.0 μg/mL)和诺氟沙星(MIC: 16 μg/mL)的2~64倍[32]。其中,杂合体18(MIC:
0.25 μg/mL)对所测的MRSA和3株金葡菌具有相同的活性,RI为1,提示该杂合体具有克服耐药性的潜力。此外,当浓度为4.0 μg/mL时,杂合体18可在4 h清除MRSA,说明该杂合体具有快速杀菌作用。作用机制研究发现,该杂合体可通过破坏细胞膜、干扰DNA复制发挥抗MRSA活性。而诺氟沙星-噻唑杂合体19(MIC: 2.0 μg/mL)的抗MRSA活性是母药诺氟沙星(MIC: 8.0 μg/mL)的4倍,且对正常BEAS-2B细胞未显示出毒性(CC50: >132 μmol/L)[33]。构-效关系显示,甲肟为高活性所必需且甲基对活性有利,将甲基用取代苯基替代将导致活性降低。作用机制研究发现,该杂合体不仅可通过干扰细胞的完整性发挥有效的膜通透性,通过氢键和π-π堆积与拓扑异构酶IV-DNA复合物结合,而且可通过插入DNA来形成稳定的生物大分子复合物,进而发挥高效抗菌活性。
环丙沙星-噻唑啉二酮杂合体20(MIC: 2.56 μg/mL)
及其衍生物21(MIC: 2.56 μg/mL)具有潜在的抗MRSA AUMC 261活性,但活性弱于母药环丙沙星(MIC: 0.20 μg/mL)[34]。构-效关系显示,向苯环无论引入供电子基还是吸电子基均不会提高抗MRSA活性。作用机制研究发现,这类杂合体可同时作用于DNA促旋酶和拓扑异构酶IV。
环丙沙星-噁二唑杂合体22(MIC: 32 μg/mL)具有中等強度的抗MRSA活性,且活性优于母药环丙沙星(MIC: 128 μg/mL)[35]。不仅如此,该杂合体(MIC: ≤0.125 μg/mL)的抗金葡菌活性是环丙沙星(MIC: 0.5 μg/mL)的≥4倍,且杀菌活性也优于环丙沙星。作用机制研究显示,该杂合体与拓扑异构酶IV具有较强的亲和力,故可能通过抑制拓扑异构酶IV发挥抗菌活性。
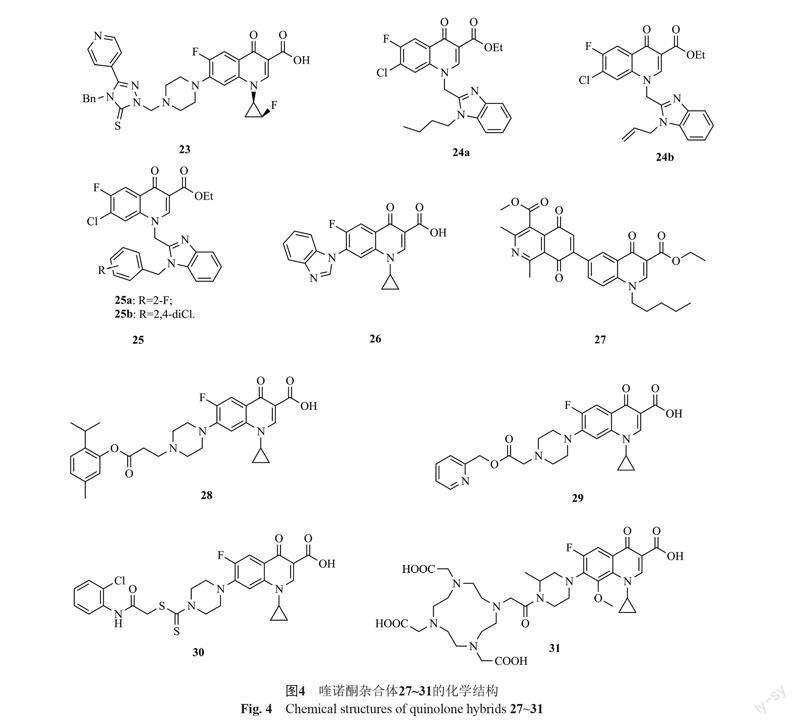
1-[(1R,2S)-2-氟环丙基]环丙沙星-1,2,4-三氮唑-5-硫酮杂合体23(图4,MIC: 50 μg/mL)也具有潜在的抗MRSA活性,但活性弱于环丙沙星(MIC: 25 μg/mL)[36-37]。构-效关系显示,1,2,4-三氮唑母核N-4位的苄基对活性有利,将其换为苯基或甲基将导致活性降低。
4-喹诺酮-苯并咪唑杂合体24a,b(MIC: 8.0 μg/mL)具有良好的抗MRSA活性,而克林沙星(MIC: >512 μg/mL)和诺氟沙星(MIC: >512 μg/mL)则未显示出任何活性[38]。构-效关系显示,苯并咪唑N-1位的烷基或烯基并非高活性所必需,向该位置引入含卤素的苄基也可耐受。其中,杂合体25a,b(MIC: 8.0 μg/mL)的活性与杂合体24a,b相当。作用机制研究发现,杂合体25a可与拓扑异构酶IV-DNA复合物相结合,抑制DNA复制,进而发挥抗菌活性。氟喹诺酮-苯并咪唑杂合体26(MIC: 0.5 μg/mL)也显示出优秀的抗MRSA活性,值得进一步研究[39]。
4-喹诺酮-异喹啉-5,8-二酮杂合体27(MIC: 8.0 μg/mL)具有潜在的抗MRSA活性,但活性弱于万古霉素(MIC: 4.0 μg/mL)[40]。作用机制研究表明,该杂合体对位于GyrA和GyrB亚单位之间的一个特定疏水口袋具有亲和力,但不与DNA分子相互作用。环丙沙星-麝香草酚杂合体28不仅对所测的4株金葡菌具有良好的活性(MIC: 0.8~1.6 μg/mL),而且对2株临床分离MRSA也具有潜在的活性(MIC: 1.0和4.0 μg/mL)[41]。作用机制研究表明,杂合体28可与拓扑异构酶IV-DNA复合物相结合,进而发挥抗菌活性。
环丙沙星-吡啶杂合体29(MIC: 12.5 μg/mL)的抗MRSA活性略弱于环丙沙星(MIC: 6.25 μg/mL)[42],而环丙沙星-二硫代氨基甲酸杂合体30(MIC: 0.097和0.39 μg/mL)的抗MSSA和MRSA活性均优于母药环丙沙星(MIC: 0.195和1.56 μg/mL)[43]。加替沙星-1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸(DOTA)杂合体31(MIC: 1.56 μg/mL)具有潜在的抗MRSA活性,但活性远逊于母药加替沙星(MIC: 0.078 μg/mL) [44]。
2 结语
喹诺酮尤其是氟喹诺酮类化合物具有广谱抗菌活性,对包括MRSA在内的多种致病菌具有良好的疗效。然而,随着这类药物的广泛使用甚至滥用,耐药性问题日趋严重。因此,开发新型抗菌药势在必行。特别值得一提的是,喹诺酮杂合体可同时作用于细菌的多个靶点,具有克服耐药性的潜力,引起了药物化学家的极大关注。近年来,药物化学家设计合成了多个系列喹诺酮杂合体,并评价了它们的体内外抗MRSA活性。此类研究丰富了构-效关系,为未来的进一步结构优化提供了一定的理论参考。
未来几年的研究可集中在:①将喹诺酮药效团与更多的抗MRSA药效团杂合,以获得更多的先导化合物,为后续研究提供量的保障;②对现有的具有潜在抗MRSA活性的喹诺酮杂合体进行深入的结构优化,进一步提升其活性,为进行体内活性评价提供更多优秀候选化合物;③对具有良好体内外抗MRSA活性的喹诺酮杂合体进行深入的临床前评价,以期有更多的候选化合物能够早日进入临床为人类健康服务。
参 考 文 献
Turner N A, Sharma-Kuinkel B K, Maskarinec S A, et al. Methicillin-resistant Staphylococcus aureus: An overview of basic and clinical research[J]. Nat Rev Microbiol, 2019, 17(4): 203-218.
Gajdács M. The continuing threat of methicillin-resistant Staphylococcus aureus[J]. Antibiotics, 2019, 8(2): e52.
Craft K M, Nguyen J M, Berg L J, et al. Methicillin-resistant Staphylococcus aureus (MRSA): Antibiotic-resistance and the biofilm phenotype[J]. MedChemComm, 2019, 10(8): 1231-1241.
Cheung G Y C, Bae J S, Otto M, et al. Pathogenicity and virulence of Staphylococcus aureus[J]. Virulence, 2021, 12(1): 547-569.
Lee A S, De Lencastre H, Garau J, et al. Methicillin-resistant Staphylococcus aureus[J]. Nat Rev Dis Primers, 2018, 4: e18033.
Tacconelli E, Carrara E, Savoldi A, et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis[J]. Lancet Infect Dis, 2018, 18(3): 318-327.
Chalmers S J, Wylam M E. Methicillin-resistant Staphylococcus aureus infection and treatment options[J]. Methods Mol Biol, 2020, 2069: 229-251.
Nandhini P, Kumar P, Mickymaray S, et al. Recent developments in methicillin-resistant Staphylococcus aureus (MRSA) treatment: A review[J]. Antibiotics, 2022, 11(5): e606.
Cong Y G, Yang S J, Rao X C. Vancomycin-resistant Staphylococcus aureus infections: A review of case updating and clinical features[J]. J Adv Res, 2020, 21: 169-176.
Liu W T, Chen E Z, Yang L, et al. Emerging resistance mechanisms for 4 types of common anti-MRSA antibiotics in Staphylococcus aureus: A comprehensive review[J]. Microb Pathogen, 2021, 156: e104915.
汪阿鵬, 冯连顺, 刘明亮, 等. 氟喹诺酮类抗菌药的最新研究进展[J]. 国外医药抗生素分册, 2019, 40(3): 171-179.
柴芸, 冯连顺, 刘明亮, 等. 喹诺酮衍生物及其抗耐甲氧西林金葡球菌活性[J]. 国外医药抗生素分册, 2019, 40(3): 200-210.
Ezelarab H A A, Abbas S H, Hassan H A, et al. Recent updates of fluoroquinolones as antibacterial agents[J]. Arch Pharm, 2018, 351(9): e1800141.
Gatadi S, Madhavi S, Chopra S, et al. Promising antibacterial agents against multidrug resistant Staphylococcus aureus[J]. Bioorg Chem, 2019, 92: e103252.
Alkhzem A H, Woodman T J, Blagbrough I S. Design and synthesis of hybrid compounds as novel drugs and medicines[J]. RSC Adv, 2022, 12(30): 19470-19484.
Kumar S H M, Herrmann L, Tsogoeva S B, et al. Structural hybridization as a facile approach to new drug candidates[J]. Bioorg Med Chem Lett, 2020, 30(23): e127514.
Lungu I A, Moldovan O L, Biris V, et al. Fluoroquinolones hybrid molecules as promising antibacterial agents in the fight against antibacterial resistance[J]. Pharmaceutics, 2022, 14(8): e1749.
Jia Y S, Zhao L Y. The antibacterial activity of fluoroquinolone derivatives: An update (2018-2021)[J]. Eur J Med Chem, 2021, 224: e113741.
Tan Y M, Li D, Li F F, et al. Pyrimidine-conjugated fluoroquinolones as new potential broad-spectrum antibacterial agents[J]. Bioorg Med Chem Lett, 2022, 73: e128885.
Song R Z, Wang Y, Wang M H, et al. Design and synthesis of novel desfluoroquinolone-aminopyrimidine hybrids as potent anti-MRSA agents with low hERG activity[J]. Bioorg Chem, 2020, 103, e104176.
Samir M, Ramadan M, Abdelrahman M H, et al. New potent ciprofloxacin-uracil conjugates as DNA gyrase and topoisomerase IV inhibitors against methicillin-resistant Staphylococcus aureus[J]. Bioorg Med Chem, 2022, 73, e117004.
Norouzbahari M, Salarinejad S, Güran M, et al. Design, synthesis, molecular docking study, and antibacterial evaluation of some new fluoroquinolone analogues bearing a quinazolinone moiety[J]. DARU J Pharm Sci, 2020, 28(2): 661-672.
Han Z S, Kirchmair J, Li X Y, et al. Discovery of N-quinazolinone-4-hydroxy-2-quinolone-3-carboxamides as DNA gyrase B-targeted antibacterial agents[J]. J Enzyme Inhib Med Chem, 2022, 37(1): 1620-1631.
Türe A, Kulaba? N, Dingi? S ?, et al. Design, synthesis and molecular modeling studies on novel moxifloxacin derivatives as potential antibacterial and antituberculosis agents[J]. Bioorg Chem, 2019, 88, e102965.
Mohammed A A M, Suaifan G A R Y, Shehadeh M B, et al. Design, synthesis, and biological evaluation of 1,8-naphthyridine glucosamine conjugates as antimicrobial agents[J]. Drug Dev Res, 2019, 80(1): 179-186.
Mohammed A A M, Okechukwu P N, Shehadeh M B, et al. Design, synthesis and antimicrobial evaluation of novel glycosylated-fluoroquinolones derivatives[J]. Eur J Med Chem, 2020, 202: e112513.
Fang B Z, Hiasa H, Lv W, et al. Design, synthesis and structure-activity relationships of novel 15-membered macrolides: Quinolone/quinoline-containing sidechains tethered to the C-6 position of azithromycin acylides[J]. Eur J Med Chem, 2020, 193: e112222.
Gao F, Ye L, Kong F, et al. Design, synthesis and antibacterial activity evaluation of moxifloxacin-amide-1,2,3-triazole-isatin hybrids[J]. Bioorg Chem 2019, 91: e103162.
Guo H. Design, synthesis, and in vitro antibacterial activities of propylene-tethered gatifloxacin-isatin hybrids[J]. J Heterocyclic Chem, 2018, 55(8): 1899-1905.
Govender H, Mocktar C, Kumalo H M, et al. Synthesis, antibacterial activity and docking studies of substituted quinolone thiosemicarbazones[J]. Phosph, Sulf Silic Rel Elem, 2019, 194(11): 1074-1081.
Xue W J, Li X Y, Ma G X, et al. N-thiadiazole-4-hydroxy-2-quinolone-3-carboxamides bearing heteroaromatic rings as novel antibacterial agents: Design, synthesis, biological evaluation and target identification[J]. Eur J Med Chem, 2020, 188, e112022.
Chen J P, Battini N, Ansari M F, et al. Membrane active 7-thiazoxime quinolones as novel DNA binding agents to decrease the genes expression and exert potent anti-methicillin-resistant Staphylococcus aureus activity[J]. Eur J Med Chem, 2021, 217, e113340.
Wang L L, Battini N, Bheemanaboina R R Y, et al. A new exploration towards aminothiazolquinolone oximes as potentially multi-targeting antibacterial agents: Design, synthesis and evaluation acting on microbes, DNA, HSA and topoisomerase IV[J]. Eur J Med Chem, 2019, 179, 166-181.
Aziz H A, El-Saghier A M M, Badr M, et al. Thiazolidine?2,4?dione?linked ciprofloxacin derivatives with broad?spectrum antibacterial, MRSA and topoisomerase inhibitory activities[J]. Mol Divers, 2022, 26: 1743-1759.
Yang P, Luo J B, Zhang L L, et al. Design, synthesis and antibacterial studies of 1,3,4-oxadiazole-fluoroquinolone hybrids and their molecular docking studies[J]. Chemistry Select, 2021, 6(46): 13209-13214.
Gao Y, Na L X, Xu Z, et al. Design, synthesis and antibacterial evaluation of 1-[(1R,2S)-2-fluorocyclopropyl]ciprofloxacin-1,2,4-triazole-5(4H)-thione hybrids[J]. Chem Biodiversity, 2018, 15(10): E1800261.
Geng Y H, Wei Z Q, Xu Z, et al. Design, synthesis and antibacterial evaluation of 1-[(1R,2S)-2-fluorocyclopropyl]ciprofloxacin-(4-methyl-3-aryl)-1,2,4-triazole-5(4H)-thione hybrids[J]. Rev Roum Chim, 2019, 64(1): 101-107.
Wang Y N, Bheemanaboina R R Y, Gao W W, et al. Discovery of benzimidazole-quinolone hybrids as new cleaving agents toward drug-resistant Pseudomonas aeruginosa DNA[J]. Chem Med Chem, 2018, 13(10): 1004-1017.
Suay-garcía B, Bueso-bordils J I, Antón-fos G, et al. Synthesis of quinolones and Zwitterionic quinolonate derivatives with broad-spectrum antibiotic activity[J]. Pharmaceuticals, 2022, 15(7): e818.
Martins F J, Sagrillo F S, Medeiros R J V, et al. Evaluation of biological activities of quinone-4-oxoquinoline derivatives against pathogens of clinical importance[J]. Curr Top Med Chem, 2022, 22: 973-991
Chaniewicz K, Kmiecik S, Koliński M, et al. Design and synthesis of menthol and thymol derived ciprofloxacin: Influence of structural modifications on the antibacterial activity and anticancer properties[J]. Int J Mol Sci, 2022, 23(12): e6600.
Shahbazi A, Mostafavi H, Zarrini G, et al. Novel N-4-piperazinyl ciprofloxacin-ester hybrids: Synthesis, biological evaluation, and molecular docking studies[J]. Russ J Gen Chem, 2020, 90(8): 1558-1565.
Esfahani E N, Mohammadi-Khanaposhtani M, Rezaei Z, et al. Biology-oriented drug synthesis (BIODS) approach towards synthesis of ciprofloxacin-dithiocarbamate hybrids and their antibacterial potential both in vitro and in silico[J]. Chem Biodiversity, 2018, 15(10): E1800273.
Li W T, Hong G, Mao L, et al. Synthesis, antibacterial evaluation and in silico study of DOTA-fluoroquinolone derivatives[J]. Med Chem Res, 2022, 31(5): 705-719.
收稿日期:2022-11-09
基金項目:河北省卫健委课题(No. 20201430)
作者简介:王林硕,女,生于1986年,工程师,主要从事新药研究,E-mail: 396809463@qq.com
*通信作者,E-mail: ohogu@qq.com
