高效液相色谱法测定乳及乳制品中唾液酸含量方法研究
2023-12-24赵梓棋李艳红卢智华张红梅毕成名王丹慧白晓玲张河霞
赵梓棋,李艳红,李 慧,卢智华,张红梅,齐 琦,毕成名,王丹慧,王 斌,白晓玲,张河霞
蒙牛乳业有限公司,内蒙古自治区 呼和浩特市 011500
0 引言
N-乙酰神经氨酸是一种重要的α-酮酸,其特点是具有9个碳的母链[1],分子结构如图1所示。在自然界,N-乙酰神经氨酸广泛分布,大多以衍生物形式存在于众多生物组织中。例如,唾液酸是N-乙酰神经氨酸的一种形式,可以在脊椎动物和哺乳动物体内广泛找到,主要分布在脑、乳、血液和神经黏液蛋白中。此外,N-乙酰神经氨酸也是组成细胞膜上糖蛋白和糖脂的关键成分[2]。

图1 N-乙酰神经氨酸的分子结构
作为一种天然的大脑营养素,N-乙酰神经氨酸积极参与多种生物学过程,包括体内的抑菌、免疫调节、抗病毒活动,以及在细胞识别和认知发育中的作用。因此,含有大量N-乙酰神经氨酸的婴幼儿产品在市场上具有明显的优势。N-乙酰神经氨酸不仅可促进婴儿大脑发育和认知能力提升[3],也有助于改善婴幼儿的肠道环境,提高对营养物质的吸收能力。因此,N-乙酰神经氨酸在婴幼儿成长过程中扮演着至关重要的角色[4]。
大部分动物组织中的唾液酸的主要形态为N-乙酰神经氨酸。在人体中,唾液酸特指N-乙酰神经氨酸。人体内的N-乙酰神经氨酸主要来源于母乳,初乳中的含量最为丰富。N-乙酰神经氨酸也存在于牛奶中。随着国民生活水平的提升,人们对食品的营养价值更加重视[5]。牛奶营养价值极高,包含丰富的矿物质和身体所需的微量元素,在预防各种慢性疾病、促进人体各项功能发育等方面效果显著。因此,将N-乙酰神经氨酸通过现代化工艺引入乳制品,将是行业的未来发展趋势。在此背景下,提升N-乙酰神经氨酸的检测技术至关重要。一种可靠、准确的N-乙酰神经氨酸检测方法可以准确确定食品的营养价值。目前已有检测N-乙酰神经氨酸含量的许多方法。如薄层色谱、硫代巴比妥酸和苯二酚比色等方法[6],这些方法具有操作简便、检测速度快、结果准确等优点[7],但受样品中糖苷和不饱和脂肪酸的影响,方法稳定性和抗干扰能力差[8]。随着科技的进步,传感器法、高效液相色谱法、液-质联用法和气-质联用法等新的检测方法不断应用[9]。其中,高效液相色谱法因其快速、便捷、定量准确且成本低等优点得到广泛应用[10]。尽管有研究已经利用高效液相色谱法测定奶粉中唾液酸的含量[11],但该方法复杂度较高,操作不易,并且不适合大批量样本的检测[12,13]。
因此,本文的目标是建立一种快速、操作简单的检测方法,以对唾液酸原样、乳制品和乳清蛋白粉中添加的N-乙酰神经氨酸进行定量分析。此检测方法的建立对于实现乳制品与N-乙酰神经氨酸的有效融合具有积极意义,可丰富相关领域的科学研究,也为生物活性物质的检测提供了数据参考。
1 材料与方法
1.1 材料与试剂
UHT奶、唾液酸原样、乳清蛋白粉;N-乙酰神经氨酸(色谱纯),上海安普实验科技股份有限公司;0.22 μm混合膜;甲醇(色谱纯)、浓硫酸、亚铁氰化钾、乙酸锌,上海麦克林生化科技股份有限公司。除另有说明外,所用试剂均为分析纯。
1.2 仪器与设备
Waters 2695高效液相色谱仪配紫外检测器,沃特世(Waters)科技有限公司;HC-3518高速离心机,科大创新股份有限公司中佳分公司;ME204E电子分析天平,梅特勒托利多仪器(上海)有限公司;SB-4200DT超声波清洗机,宁波新芝生物科技股份有限公司;移液器,赛默飞世尔科技(中国)有限公司;实验用水按照GB/T 6682 规定的一级实验用水。
1.3 实验方法
1.3.1 溶液的配制
N-乙酰神经氨酸标准溶液(10 mg/mL):准确称取0.100 0 g(精确至0.000 1 g)N-乙酰神经氨酸标准品,用一级水溶解,转移至容量瓶中,加水定容至10 mL,该溶液不能长期保存,需现用现配。
0.005 mol/L 硫酸水溶液:准确吸取0.267 mL硫酸注入少量一级水中,用一级水稀释至1 L,用0.22 μm滤膜过滤,经超声脱气10 min后备用。
0.005 mol/L 硫酸甲醇溶液:准确吸取0.268 mL硫酸注入少量甲醇中,用甲醇稀释至1 L,用0.22 μm滤膜过滤,经超声脱气10 min后备用。
1.3.2 样品的制备
(1)UHT奶试样处理
准确称取0.200 0 g(精确到0.000 1 g)样品于100 mL容量瓶中,加入约50 mL水,加1 mL的106 g/L亚铁氰化钾溶液,摇匀,加1 mL的92 g/L乙酸锌溶液摇匀,加水定容,摇匀。取部分溶液9 000 r/min离心10 min,样液经0.22 μm滤膜过滤,滤液待测定。
(2) 唾液酸原样处理
准确称取0.200 0 g(精确到0.000 1 g)样品于烧杯中,用少量水溶解后转移至100 mL容量瓶中,加水定容,摇匀。取部分溶液9 000 r/min离心10 min,样液经0.22 μm滤膜过滤,滤液待测定。
(3) 乳清蛋白粉试样处理
准确称取0.200 0 g(精确到0.000 1 g)样品于烧杯中,用少量水溶解后转移至100 mL容量瓶中,加1 mL的106 g/L亚铁氰化钾溶液摇匀,加1 mL的92 g/L乙酸锌溶液摇匀,加水定容,摇匀。取部分溶液9 000 r/min离心10 min,样液经0.22 μm滤膜过滤,滤液待测定。
1.4 色谱条件
色谱柱:C18色谱柱(250 mm×4.6 mm,5 μm),配有柱温箱。流动相A:0.005 mol/L 硫酸水溶液。流动相B:0.005 mol/L 硫酸甲醇溶液,梯度洗脱,紫外检测波长为210 nm,进样体积10 μL,优化后的梯度洗脱程序见表1。
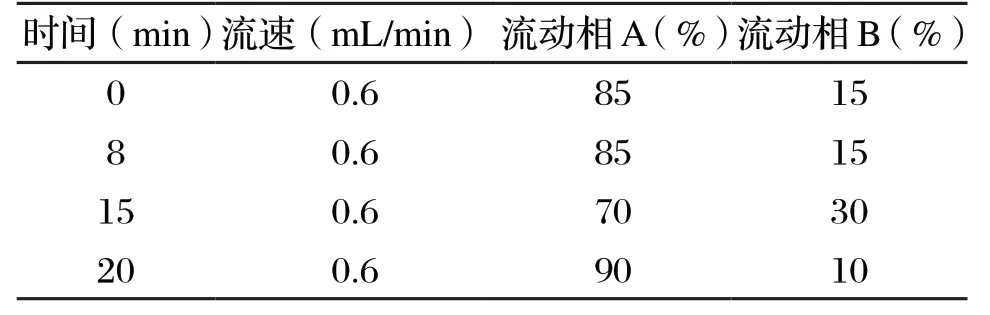
表1 梯度洗脱程序
1.5 标准曲线制作
分别准确量取标准溶液0、0.2、0.5、1、1.5、2、3 mL于容量瓶中,加水定容至10 mL(浓度为0、0.2、0.5、1.0、1.5、2.0、3.0 mg/mL)。取上述各标准溶液10 μL进样。以N-乙酰神经氨酸浓度为横坐标,峰面积为纵坐标绘制标准曲线。
1.6 单因素和正交实验设计
1.6.1 单因素实验设计
设置固定条件,采用梯度洗脱法中使用0.005 mol/L的硫酸溶液和0.005 mol/L的硫酸甲醇溶液作为流动相。以出峰保留时间为评价指标,分别以柱温(30、35、40、45 ℃)、流动相流速(0.5、0.6、0.7、0.8 mL/min)、流动相pH值(1.0、2.0、3.0、4.0)、流动相比例(75∶25、80∶20、85∶15、90∶10)等四个因素进行单因素实验。
1.6.2 正交实验设计
根据单因素实验结果,对柱温(A)、流动相流速(B)、流动相pH值(C)、流动相比例(D)进行四因素三水平正交实验,优化高效液相色谱法检测乳及乳制品中的唾液酸含量最佳实验条件,正交因素水平表见表2。
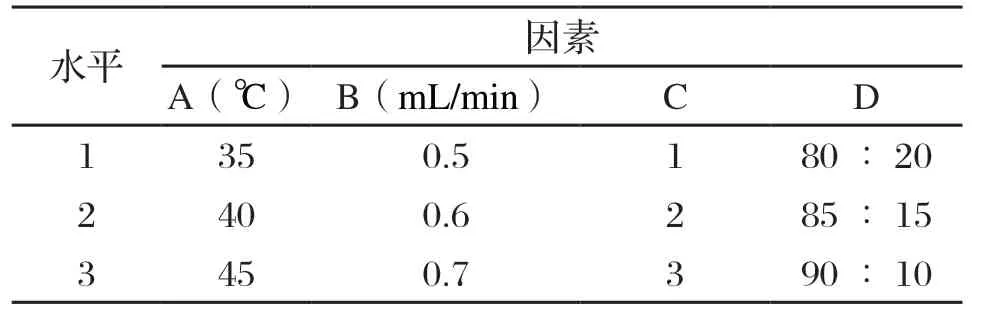
表2 正交因素水平表
1.7 色谱分析结果表述
试样中N-乙酰神经氨酸含量按公式(1)进行计算:
式中:
X——试样中N-乙酰神经氨酸含量,单位g/100g;
C——进样样液中 N-乙酰神经氨酸含量,单位mg/mL;
V——样品定容体积,单位mL;
m——样品取样量,单位g;
计算结果保留3位有效数字。
2 结果与分析
2.1 单因素实验结果分析
N-乙酰神经氨酸具有紫外吸收能力,本实验在紫外检测器210 nm处配合C18色谱柱对其进行分析。影响N-乙酰神经氨酸出峰的主要因素有流动相pH值、流动相比例、流动相流速、柱温[14]。因此我们通过寻找最佳的色谱条件流动相pH值、流动相比例、流动相流速、柱温,对N-乙酰神经氨酸的检测工艺进行优化。
2.1.1 流动相流速的选择
流动相的流速是影响N-乙酰神经氨酸保留时间的重要因素。当流速增大时,流动相洗脱能力增强,保留时间变小,峰形较窄,实验时间短,同一时间处理样品效率高[15];当流动相流速减小时,流动相洗脱能力降低,保留时间增加,峰形更宽,同一时间处理样品效率降低[16]。本文主要研究样品在流动相流速为0.5 mL/min、0.6 mL/min、0.7 mL/min、0.8 mL/min时对保留时间的影响。如图2所示,样品中N-乙酰神经氨酸检测出峰完整,无包峰,杂峰较少。流动相流速改变时,样品峰形变化差异较小,保留时间变化差异较大,当流动相流速为0.5 mL/min、0.6 mL/min、0.7 mL/min、0.8 mL/min时,保留时间分别为6.23 min、5.69 min、5.58min、5.03 min。流动相流速较低时样品分析时间较长,分析效率较低,流动相流速较大时,液相系统压力较大,对管路和色谱柱损伤较大,综合考虑选择流速为0.6 mL/min进行实验。
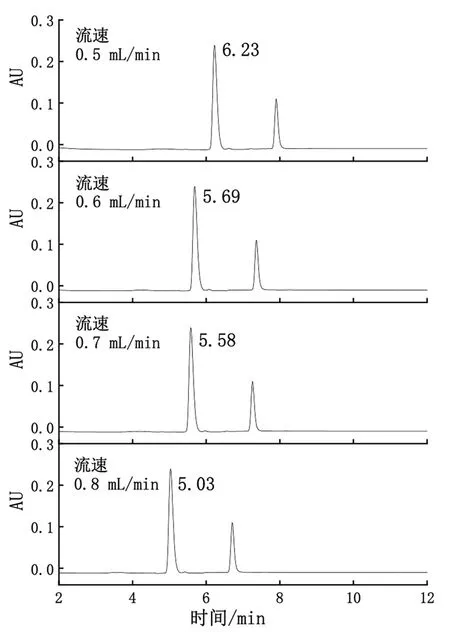
图2 流动相不同流速下样品色谱峰图
2.1.2 柱温的选择
高效液相色谱法检测法时,柱子温度对保留时间和色谱峰形有一定影响,通过检测样品中物质与固定相的结合-分离差异产生分离作用,温度对物质的结合-分离过程产生影响,进而影响保留时间和色谱峰形[17]。因此本文对色谱柱的温度进行优化,以保留时间和色谱峰形为评价指标,分别在柱子温度为30 ℃、35 ℃、40 ℃、45 ℃时进样。当柱子温度较高时,物质的结合-分离过程加快,但是样品的保留时间不稳定,会加大分析的复杂程度,物质的分辨率降低[18];如果柱子温度较低,会延长分离过程,分辨率增高,低温下流动相的粘度相对较高,较长的保留时间会增加蠕动泵和色谱柱的损伤[19]。如图3所示,分别在色谱柱温30 ℃、35 ℃、40 ℃、45 ℃下检测,色谱峰峰形基本相似,无前倾、拖尾;色谱柱温为30 ℃、35 ℃、40 ℃、45 ℃时,保留时间分别为6.20 min、5.93 min、5.68 min、5.28 min。样品中唾液酸色谱峰均能够达到很好的分离,样品中其它杂质峰对主峰测定无干扰[20]。随着色谱柱温度的增加,保留时间缩短。所以在柱温为45 ℃时保留时间最小,但柱温较高时分离-结合的不稳定性加大;温度较低时洗脱效率较低,时间更长,增大色谱柱的损伤[21]。综合考虑温度的影响和分离效果以及效率的情况下,选择柱温为40 ℃进行实验。
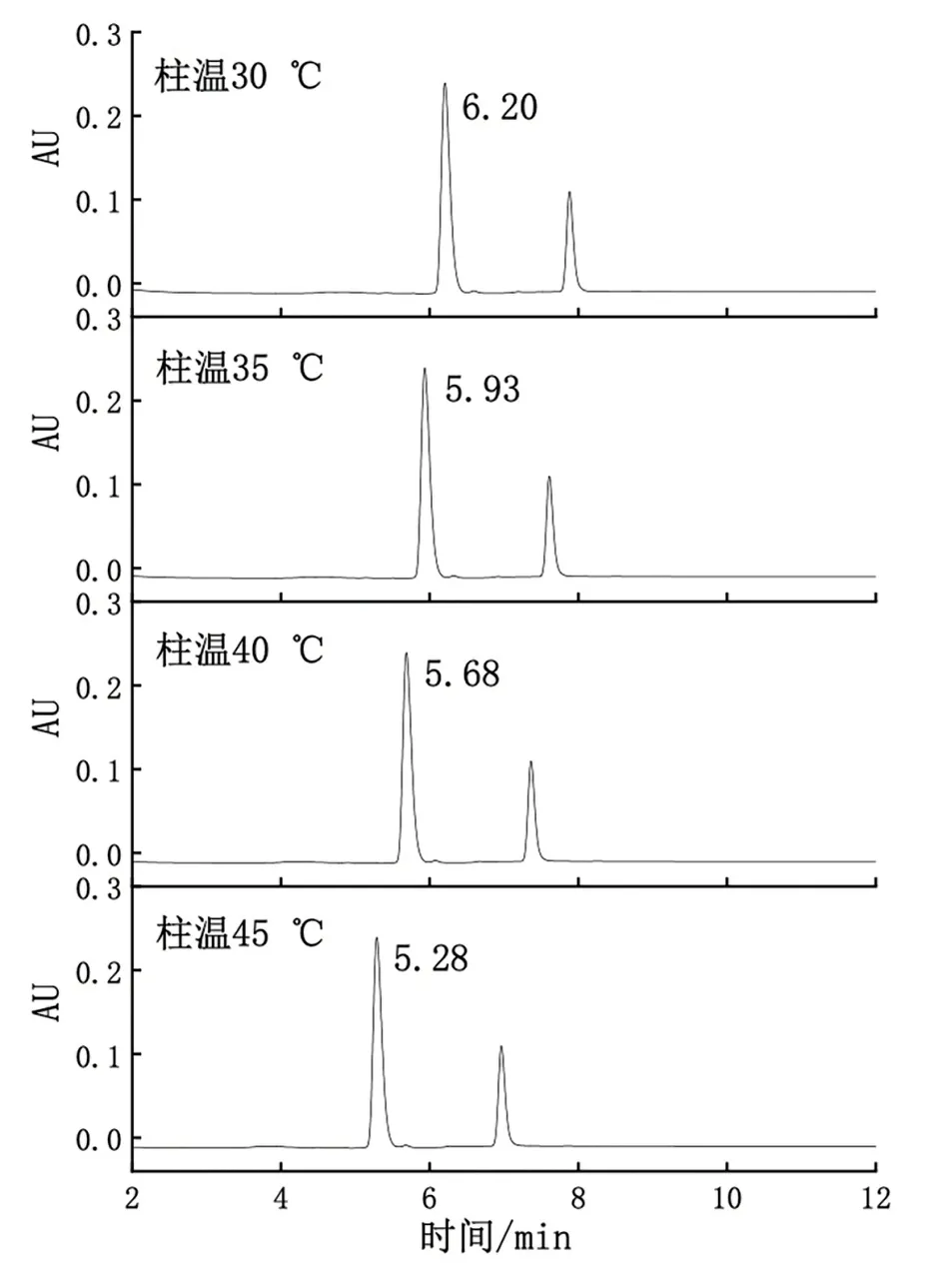
图3 不同色谱柱温下样品色谱峰图
2.1.3 流动相比例的选择
流动相所用溶剂和比例的变化影响流动相的极性,对溶剂的洗脱能力和强度产生影响[22],进而影响物质的保留时间和响应值[23]。本文采用0.005 mol/L硫酸水溶液和0.005 mol/L硫酸甲醇溶液为流动相,以保留时间和峰形为指标对流动相的比例进行优化,硫酸水溶液和硫酸甲醇溶液的体积比分别为90∶10、85∶15、80∶20、75∶25(V/V)对色谱峰分离的影响。如图4所示,不同流动相比例对保留时间和峰形有较大的影响,随着有机相比例的增加,洗脱能力和洗脱效率提高,保留时间逐渐缩小,色谱峰形变化不明显。水相:有机相比例为90∶10(V/V)时流动相洗脱能力降低,保留时间最大为6.58 min。保证减少仪器设备损伤和节约药品以及分离效果的基础上,综合考虑选择流动相水相:有机相比例为85∶15(V/V)时进行实验。
2.1.4 流动相中水相pH值的选择
流动相的pH值对色谱峰的峰形产生较大影响,本文通过加入硫酸来调整流动相的pH值,抑制其解离速度,改变极性可解离化合物的容量因子,提高分离选择性和色谱峰的对称性,研究pH值对峰形和保留时间的影响[24]。本文采用硫酸调整水相pH值为1.0、2.0、3.0、4.0进行分析。如图5所示,所有样品出峰完整,杂峰较少,不影响N-乙酰神经氨酸色谱峰的分析计算。流动相pH值为1和4时保留时间相差0.1分钟,pH值差异对保留时间有影响,pH值为1时色谱峰形有一定的前延、不对称,并且pH值较低时,对C18色谱柱损伤较大,不利于长期大量检测。说明改变pH值对保留时间和峰形有一定影响。表明在流动相水相:有机相比例为85∶15(V/V)的体系中,pH值相差较小时对N-乙酰神经氨酸与流动相的分离与结合影响较小。综合考虑到经济型和分离效果,选择pH值为2进行实验。

图5 不同流动相pH 值下样品色谱峰图
2.2 正交实验结果分析
2.2.1 正交实验设计与结果分析
正交实验设计与结果如表3所示,影响唾液酸保留时间的因素主次顺序为:A、B、D、C,实验组A3B3C2D1的保留时间最短为5.42 min,A2B1C2D3的保留时间最长为5.79 min。理论保存时间最短组合为A3B3C1D1,最长组合为A1B1C3D3。以实验效率较快和仪器损伤较低的角度考虑,选择保留时间长短适中的实验组为A2B2C3D1,理论组为A2B2C2D2,理论组合实验组条件不同,进行验证性实验确定最佳实验条件。

表3 正交实验设计与结果
2.2.2 验证性实验
通过正交实验结果分析,对A2B2C2D2进行验证性实验,重复三次。唾液酸保留时间为5.69 min,与实验组5保留时间几乎相同,结合样品前处理和实验5流动相pH值为1,对色谱柱损伤较大,选择理论组合A2B2C2D2为最佳实验条件。所以检测唾液酸含量的最佳实验条件为:柱温为40 ℃、流速为0.6 mL/min、流动相pH值为2.0、流动相比例为85∶15。
2.3 N-乙酰神经氨酸色谱法分析方法学考察
2.3.1 标准曲线回归方程、线性范围、相关系数、检出限及定量限分析
分别准确量取标准溶液0、0.2、0.5、1、1.5、2、3 mL于容量瓶中,加水定容至10 mL(浓度为0、0.2、0.5、1.0、1.5、2.0、3.0 mg/mL)。取上述各标准溶液10 μL进样检测。以N-乙酰神经氨酸浓度为横坐标,峰面积为纵坐标绘制标准曲线,所得线性回归方程和相关系数见表4。相关系数为0.998 944,标准方程线性范围为0~3.0 mg/mL,标准曲线方程结果满足后续实验分析要求。如图6所示,N-乙酰神经氨酸标准品保留时间为5.69 min,色谱峰图出峰完整,峰形对称,无拖尾,杂峰较少[25]。表明此方法检测3种样品中的N-乙酰神经氨酸含量结果较为准确。

表4 N-乙酰神经氨酸线性方程、线性范围、相关系数、检出限及定量限实验结果

图6 N-乙酰神经氨酸标准品色谱峰图
2.3.2 精密度实验
取液态奶样品加标量为25 g/100 g、50 g/100 g、100 g/100 g和唾液酸原样以及乳清蛋白粉加标量为50 g/100 g的样品分别重复进样6次,测定N-乙酰神经氨酸含量,并计算平均值和标准偏差。精密度结果如表5所示,液态奶样品中N-乙酰神经氨酸检测精密度为0.54%~2.59%,表明此方法检测样品中N-乙酰神经氨酸精密度良好。
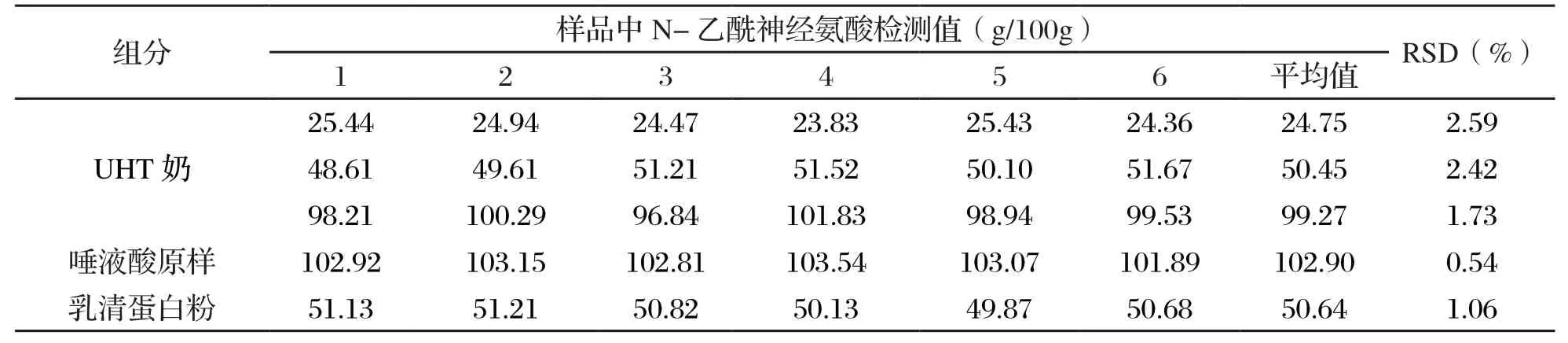
表5 N-乙酰神经氨酸的精密度实验结果
2.3.3 重现性实验
取3种样品分别经前处理后过滤到6个进样瓶,进行检测。检测3种样品中N-乙酰神经氨酸含量并计算相对应的平均值和标准偏差,计算结果如表6所示,3种样品的RSD值分别为0.44%、0.27%、0.95%,表明该方法检测3种样品中N-乙酰神经氨酸重现性较好,满足实验检测分析要求。

表6 样品中N-乙酰神经氨酸重现性实验结果
2.3.4 回收率实验
采用加标回收法,分别取UHT奶、唾液酸原样、乳清蛋白粉向样品中分别加入定量的N-乙酰神经氨酸进行检测,每个样本平行3次实验,计算各样本的回收率和不同样本的平均回收率以及标准偏差。如表7所示,液态奶和乳清蛋白粉中没有检测出,唾液酸原样中检测乙酰神经氨酸平均含量为108.3 g/100 g,加标法检测回收率,液态奶回收率在99.20%~101.60%,唾液酸原样在99.80%~103.94%,乳清蛋白粉在100.40%~103.66%,平均回收率分别为100.82%、102.12%、102.48%,回收率均符合实验要求;RSD分别为1.08%、1.59%、1.51%,表明此方法检测3种样品中的N-乙酰神经氨酸含量数据准确可靠。

表7 N-乙酰神经氨酸的回收率实验结果
3 结论
在本研究中,成功地建立了一种基于高效液相色谱-紫外检测器的方法,用于测定唾液酸原样、乳制品和乳清蛋白粉中添加的的N-乙酰神经氨酸含量。所设计的前处理步骤通过使用亚铁氰化钾溶液和乙酸锌溶液有效地沉淀了乳制品中的蛋白质,从而克服了乳制品中其他物质的干扰。经过在8 000 rpm/min的条件下离心10分钟,再通过0.22 μm滤膜过滤,样本即可进行检测。同时根据保留时间和色谱峰形作为评价指标,对流动相流速、比例、pH值和色谱柱温度进行了优化发现选用C18色谱柱、210 nm的紫外检测器检测波长、40 ℃的色谱柱温度、pH值为2的流动相、0.6 mL/min的流速,以及85∶15的流动相水相与有机相比例,并使用梯度洗脱可得出最优结果。此外,还对此检测方法的精密度、重复性和回收率进行了评估。本研究所建立的方法具有良好的重复性、精密度高、回收率符合、检测时间短、检测过程简单便捷等优点。适用于唾液酸原样、乳制品、乳清蛋白粉中进行N-乙酰神经氨酸含量的测定,此方法的成功开发为含N-乙酰神经氨酸乳制品的开发提供有力支持,对乳制品行业的多元化发展具有重要意义。
