钙循环碳酸化过程CaO 催化CO 脱除NO 的DFT 研究
2023-12-11初雷哲柴守冰李英杰
初雷哲 , 柴守冰 , 李英杰
(1.水发集团有限公司, 山东 济南 250101;2.山东环保产业研究院有限公司, 山东 济南 250100;3.山东大学 能源与动力工程学院, 山东 济南250061)
长期以来,由于化石燃料的广泛应用,CO2排放量急剧增加。温室效应导致全球变暖已成为人类社会的一个重大问题[1]。因此,燃煤发电厂CO2排放控制技术日益重要。钙循环(CaL)技术因其成本低、效率高等优点成为大规模CO2捕集最有前景的技术之一[2-3]。CaL 技术通过煅烧/碳酸化循环实现CO2的捕集[4-5],反应方程式为
首先,钙基吸收剂在煅烧炉中煅烧( > 900 ℃),分解为CaO 和CO2。然后,CaO 进入碳酸化炉吸收烟气中的CO2生成CaCO3(600~700 ℃)。生成的CaCO3进行下一次煅烧/碳酸化循环[6]。
燃煤发电厂也是NOx排放的主要来源之一[7]。目前,以NH3为还原剂的NH3-SCR 是电厂应用的主要技术。然而,氨泄漏和高成本是NH3-SCR 的主要缺点[8]。与NH3相比,CO 具有成本低、毒性小等优点[9-10]。LIAO 等[11]在流化床(600~1 000 ℃)中,进行了CaO 催化CO 还原NO 的测试,发现NO 的脱除效率约为75%。因此,CaO 是CO 还原NO 合适的催化剂。
煅烧炉需要煤或生物质的燃烧提供CaCO3分解所需热量[12]。其中一些未燃尽的焦炭不可避免地随CaO 进入到碳酸化炉中,被烟气中的O2氧化生成CO[13]。GAO 等[14]发现,碳酸化炉中焦炭氧化产生的CO 体积分数高达1.2%。同时,钙循环碳酸化阶段存在大量的CaO。因此,碳酸化炉中CaO 可以催化CO还原NO。SHIMIZU 等[15]在碳酸化炉中测试了焦炭的NO 还原性能。在CaO 的催化下,焦炭氧化生成的CO 还原NO 的效率为25%。ZHANG 等[16]使用鼓泡流化床反应器测试了CaL 碳酸化阶段CO 脱除NO 的能力,发现CO2/NO 脱除效率可达90%以上。然而,关于CaO 在碳酸化阶段催化CO 还原NO 的微观反应机理尚不清楚。
密度泛函理论(Density Functional Theory,DFT)广泛应用于气固反应机理的研究。通过研究原子的电子特性、几何参数等,对微观的机理进行探究,从而对实验结果进行补充和拓展。YANG 等[17]采用DFT在钙循环中测试了钙基吸收剂协同捕集SO2和CO2的性能,发现水蒸气在吸附过程中起到了增强碳酸化的作用。HE 等[18]通过DFT 研究了钙循环过程中CaCO3分解在原子水平上的详细反应路径,发现由于H2O 的存在,反应速率显著增加。OU 等[19]通过DFT计算系统地研究了Pt/Ni(111)表面上CO2加氢的完整反应路径,确定了活性位点和反应机理。Pt 在Ni表面的掺杂可以促进CO2转化为化 CO,并降低了H2解离中的阻力。WANG 等[20]利用DFT 研究了钙循环过程中,H2存在时CaO 将捕获的CO2原位转化为CO 的反应机理; C加氢是该反应中的速率控制步骤,能垒为3.12 eV。
烟气中存在水蒸气会使钙基材料的碳酸化转化率提升[21],进而影响CO 和NO 在表面的吸附。此外,SO2能与CaO 反应生成CaSO4,对CO 和NO 的吸附产生不利影响,但碳酸化反应器中的钙硫摩尔比非常大(通常在200∶1 以上)[7,22],因此SO2的影响可能较小,有必要进一步研究。
为了探究钙循环碳酸化阶段CaO 对CO 还原NO 性能的影响,笔者利用DFT 研究了CaL 碳酸化阶段CaO 催化CO 还原NO 的反应机理,计算了CO 和NO 在CaO 表面的吸附能和态密度,确定了该过程的详细反应路径,分析了各基元反应的能垒,为燃煤电站CO2和NOx的协同脱除提供理论指导。
1 计 算
1.1 计算方法
计算采用DFT 方法,采用广义梯度近似(Generalized Gradient Approximation,GGA)和Perdewburke-Ernzerhof(PBE)波函数来获得交换的相关能量。在CaO 表面吸附的几何优化计算过程中,能量、原子间作用力和最大位移的阈值分别为2.63×10-2kJ/mol、52.51 kJ/(mol·nm)和0.5×10-3nm。布里渊区的k点表示波矢空间中的离散位置,取样网格采用monkhorst-pack 方法生成,密度为2×2×1。考虑到整个计算期间的计算精度和效率,SCF 计算应用费米涂抹来加速收敛,涂抹值设置为0.1 eV[23]。为了得到准确的结果和节省计算时间,轨道截止值应大于系统中所有元素的最大值。本计算所涉及的4 个元素(O、C、Ca、N)的轨道截止值分别为0.33、0.37、0.55 和0.34 nm。因此,在计算过程中选择0.56 nm 作为轨道截止值。利用LST/QST(线性同步变换/二次同步变换)工具,研究了CaL 碳酸化阶段,CaO 催化CO 还原NO 反应的过渡状态(TS)。为了确定TS 的真实性,基于伽马点动力学矩阵和配分函数计算了TS 结构的振动频率,确保每个TS 结构只观察到一个虚频率[24]。CaO(100)表面是具有低表面能的低指数平面。很多文献报道了CO2、CO 和NO 在纯CaO(100)和掺杂CaO(100)表面的吸附[12,17,25]。因此,从CaO 晶胞出发,建立了CaO(100)周期性模型。此外,CaO(100)表面的2×2 超晶胞足以反映CaO 的表面特性,已用于开展H2O[26]和CO2[27]在CaO(100)表面的吸附研究。超晶胞越大,计算精度越高,但计算量也随之增加。综合考虑本文利用周期性边界条件创建了具有2×2 超晶胞的3 层CaO(100)表面。其中,底层采用分数坐标固定,顶部2 层和吸附质允许松弛。
1.2 能量计算
吸附能的计算方法为
其中,Ead为CaO 表面上吸附质的吸附能,eV;Etotal为整个构型吸附后的总能量,eV;EA为吸附前吸附质的能量,eV;Esurface为CaO 表面模型的能量,eV。吸附能越小,表面和吸附质之间的相互作用越强[28]。
反应路径中的不同基质分别用反应物(R)、产物(P)、中间体(IM)及过渡态(TS)表示。反应能垒计算方法为
式中,Ebar为反应能垒,eV;ETS为过渡态的能量,eV;ER为反应物的能量,eV。
2 结果与讨论
2.1 CaO(100)表面结构
CaO(100)表面优化模型的正视图和俯视图如图1 所示。为了保证在CaO(100)表面上吸附的CO和NO 是最稳定的结构,本工作考虑了CaO(100)表面上几种可能的吸附位,如图1(b)所示。可能的吸附位置有:O-top 位、Ca-top 位、Bridge 位和Hollow 位。为了验证CO、NO 的最优吸附位置,分别将分子以0.3 nm 的距离平行放置在CaO(100)表面4 个吸附位置上方。
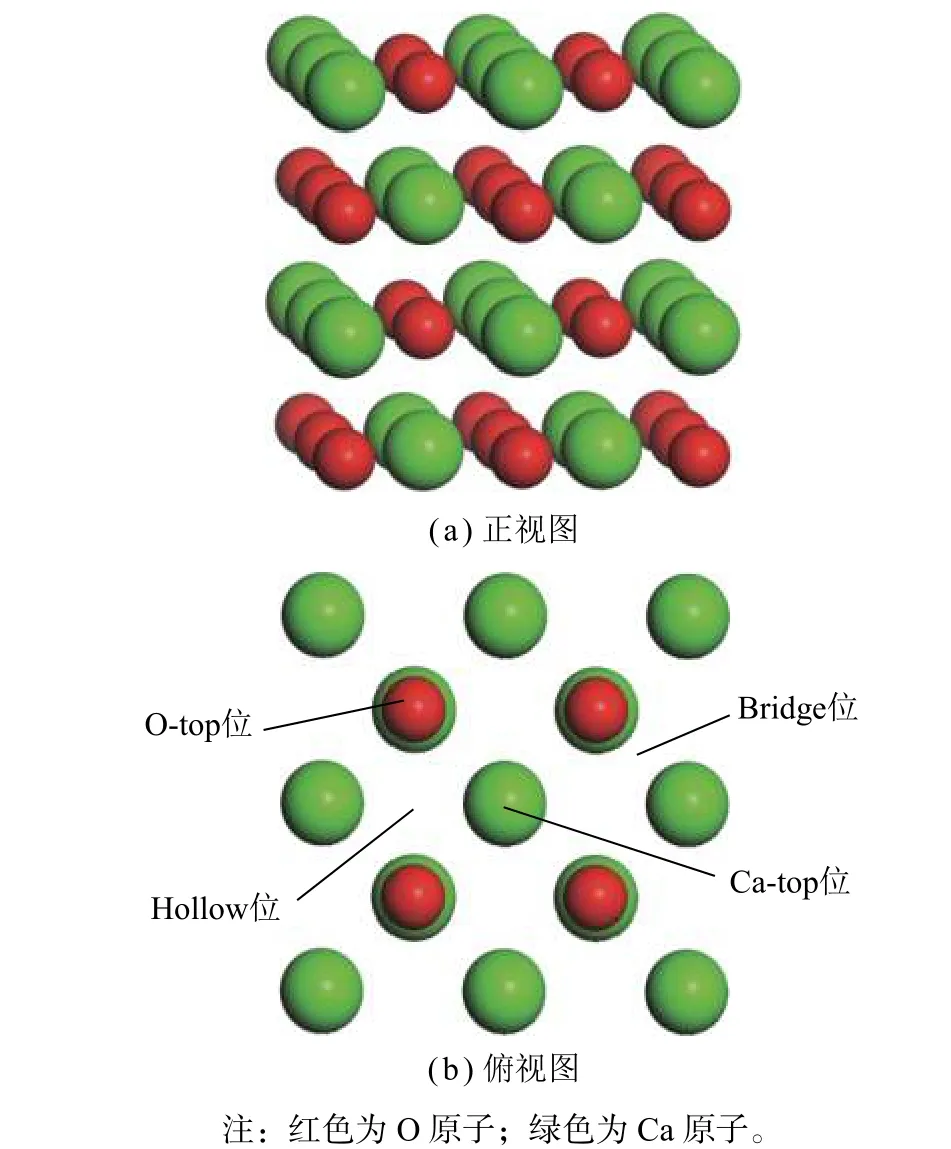
图1 CaO(100)表面优化结构的正视图和俯视图Fig.1 Side and top views of the optimized structure for CaO(100) surface
2.2 CaO 吸附CO 特性
有研究已经表明O-top 位是CO2在CaO(100)表面的最佳吸附位[29]。因此,笔者研究CO 和NO 分子在CaO(100)表面上最稳定的吸附结构。
图2 为CO 在CaO(100)表面上潜在吸附位置的优化构型。CO 的结构参数和吸附能见表1。CO 在各吸附位置的吸附能均为负值,说明CO 在CaO(100)表面是弱化学吸附。通过比较表1 中的数据,发现吸附能按照从大到小的顺序排列为:Ca-top 位 > Hollow位 > Bridge 位 > O-top 位。此外,结构参数(dC-Os)也遵循同样的规律,Ca-top 位 > Hollow 位 > Bridge 位 >O-top 位。综上所述,CO 分子在O-top 位的Ead,CO和dC-Os均为最小值,O-top 位是其在CaO (100)表面最稳定的吸附位。所以,图1(a)所示构型在接下来的计算中被选为最合适构型。
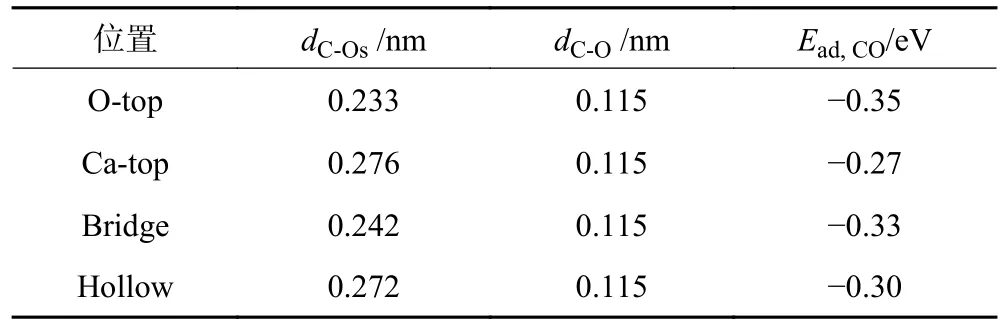
表1 CaO(100)表面对CO 的吸附能和结构参数Table 1 Adsorption energies and structural parameters of CO on CaO(100) surface
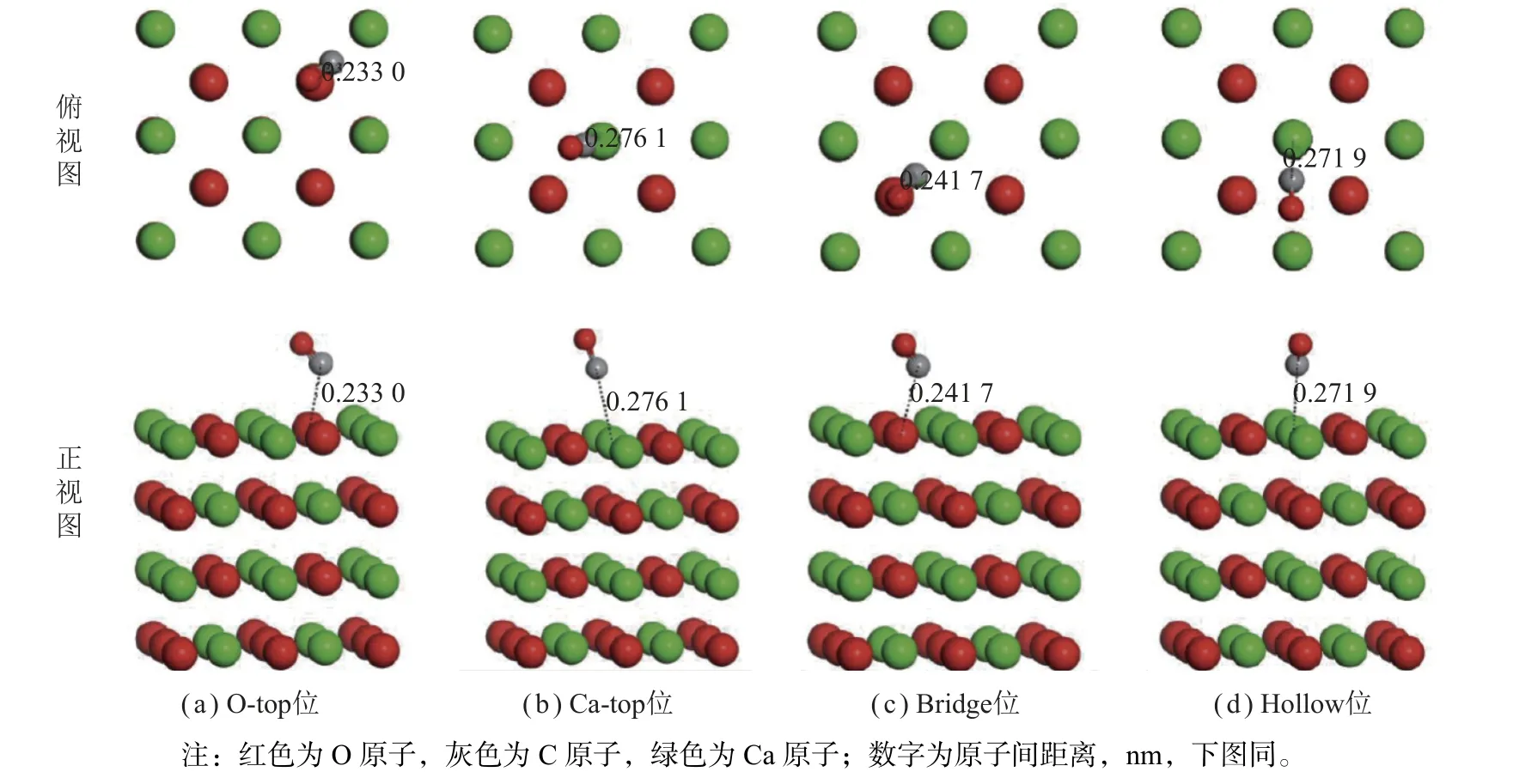
图2 CaO(100)表面CO 吸附的优化结构Fig.2 Optimized structures for CO adsorption on CaO(100) surface
为了研究吸附对原子间相互作用和电子分布变化的影响,利用分态密度(Pre-Departure Orientation Seminar,PDOS)分析了O-top 位吸附前后CO 中C 原子和CaO 中O 原子的电子状态,如图3 所示。
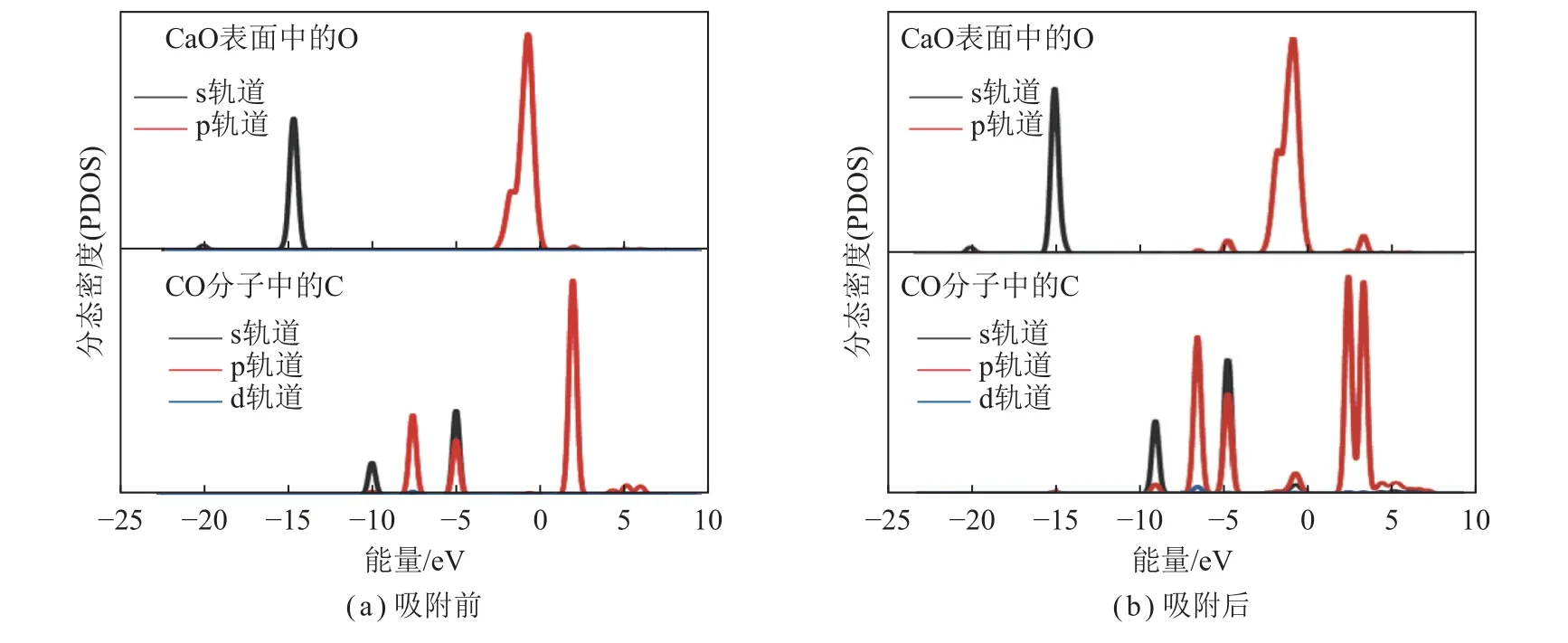
图3 CaO(100)表面吸附CO 前后的PDOSFig.3 PDOS for CaO(100) surface before and after CO adsorption
在吸附之前,如图3(a)所示,由于CO 中C 原子和CaO 中的O 原子之间没有相互作用,所以PDOS没有发生共振。从图3(b)可以看出,当CO 吸附在CaO(100)表面后,CO 中C 原子和CaO 中的O 原子的峰出现了明显的重叠。这表明CaO(100)表面的O和C 原子之间形成了共价键。原子之间重叠的电子轨道越多[30],形成的键越稳定。因此,CaO(100)表面为CO 提供了稳定的吸附位点,这有利于之后其催化CO 还原NO。
2.3 CaO 吸附NO 特性
CaO(100)表面吸附NO 的最稳定结构如图4 所示,计算得到的相应结构参数和吸附能见表2。根据图4 和表2 可以断定NO 在CaO(100)表面的吸附位点位上形成了稳定的结构,表明NO 分子在这些位点上发生了弱化学吸附。CaO(100)表面的O-top位吸附NO 的吸附能为-0.79 eV,是所有潜在吸附位中的最小值,这表明NO 更倾向于吸附在CaO(100)表面的O-top 位上。因此,在进一步研究中选择Otop 位作为最佳吸附位。同时对比表1、2,发现NO在CaO(100)表面各个吸附位点的吸附能均小于CO,这表明NO 与CaO 之间的相互作用强于CO 与CaO之间的相互作用,NO 更容易吸附在CaO 上。
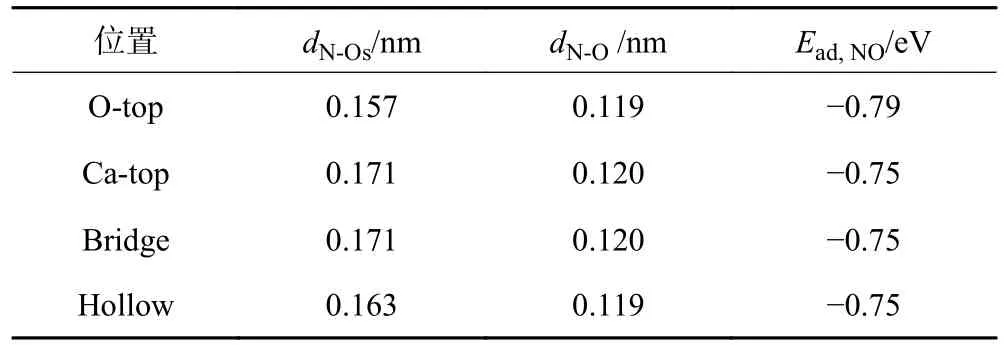
表2 CaO(100)表面对NO 的吸附能和结构参数Table 2 Adsorption energies and structural parameters of NO on CaO(100) surface
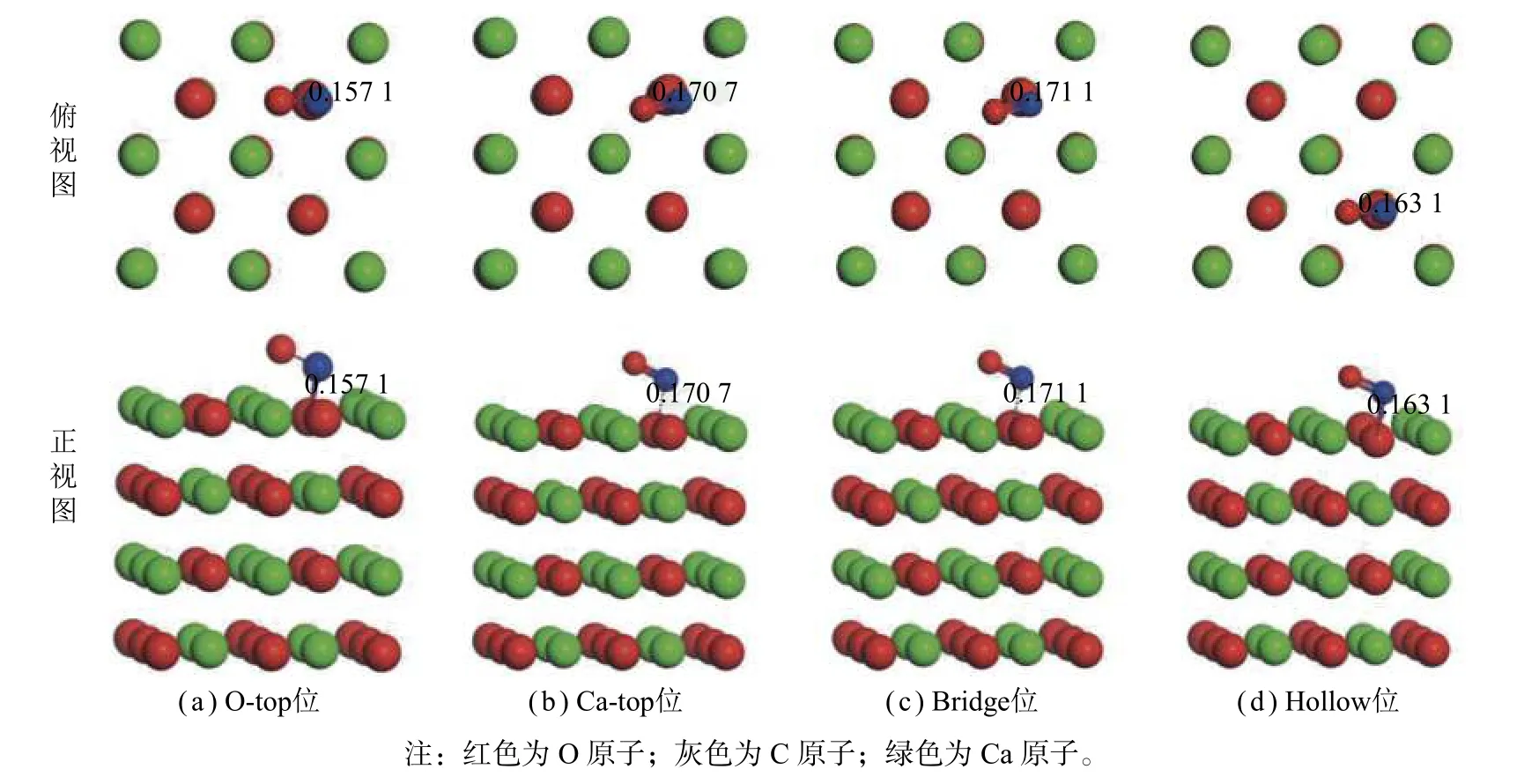
图4 CaO(100)表面NO 吸附的优化结构Fig.4 Optimized structures for NO adsorption on CaO(100) surface
NO 吸附CaO(100)表面前后的PDOS 图(图5)揭示了最佳构型中所选原子的电子状态。考虑到Otop 位是CaO 吸附NO 最合适的吸附位,因此仅对Otop 位吸附NO 进行PDOS 计算。从图5(b)可以观察到CaO(100)表面O 原子的s 轨道的峰在吸附NO 后向低能量方向移动,表明NO 的化学吸附和生成产物的高稳定性[25]。此外,吸附NO 后,CaO(100)表面O原子与NO 中N 原子轨道出现更明显的杂化现象,产生稳定的共价键。因此,CaO(100)表面对NO 的化学吸附作用强, 这有利于后续催化CO 脱硝。
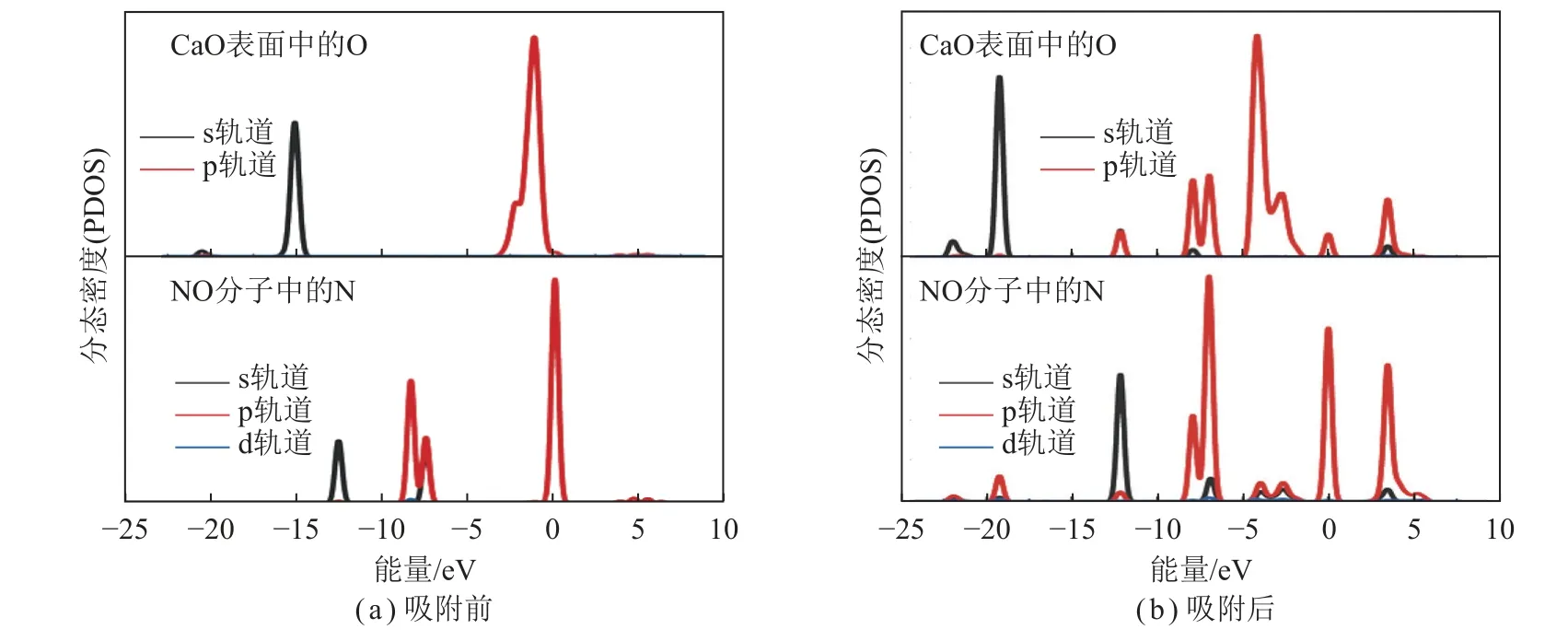
图5 CaO(100)表面吸附NO 前后的PDOSFig.5 PDOS for CaO(100) surface before and after NO adsorption
2.4 CaO 共吸附CO2、CO 及NO 特性
为了分析CO2、CO 和NO 分子在CaO(100)表面的吸附特性,将CO2、CO 和NO 分子分别吸附在CaO(100)表面的最佳吸附位上。CaO(100)表面共吸附CO2、CO 和NO 的优化结构如图6 所示。CO2、CO 和NO 分子在CaO(100)表面共吸附时的吸附能分别为Eco-ad,CO= -0.36 eV,Eco-ad,NO= -0.20 eV,其中,Eco-ad,CO和Eco-ad,NO分别为分子共同吸附在CaO(100)表面时CO、NO 的吸附能。如图6 所示,CO2分子以CO离子的形式吸附在CaO(100)表面O-top 位上。
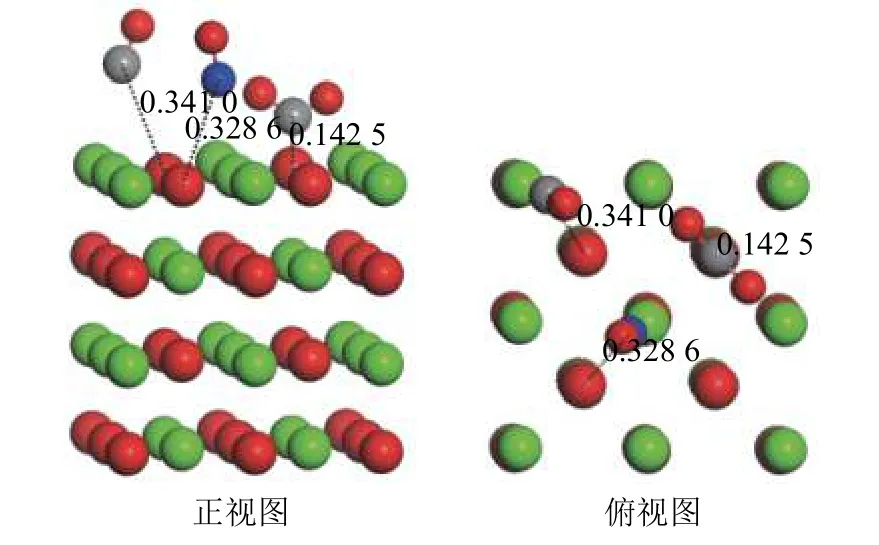
图6 CO2、CO 和NO 分子在CaO(100)表面的共吸附优化结构Fig.6 Optimized structure of CO2, CO and NO co-adsorbed on CaO (100) surface
结合表1、2 可知,在CaO(100)表面,Eco-ad,NO比Ead,NO高约75%,而Eco-ad,CO与Ead,CO相似。这表明在共吸附过程中,CO2与NO 竞争CaO(100)表面的Otop 位,从而抑制了NO 在表面的吸附。这从原子层面解释了ZHANG 等[16]工作中CO2存在时,CaO 的催化活性急剧下降的原因。
2.5 CaO 催化CO 还原NO 反应的过渡态和能垒
钙循环碳酸化阶段CaO(100)表面CO 还原NO的反应过程涉及的各驻点结构如图7 所示,能量变化见表3。由图7 可知,CaO 催化CO 还原NO 反应过程包括4 个过渡态和5 个中间体,最终生成3 个CO离子和1 个N2分子。根据反应中分子的变化,整个反应过程主要包括CO2形成与吸附、N2形成这2 个基元反应阶段。

表3 碳酸化阶段CaO(100)表面CO 还原NO 的反应能垒Table 3 Ebar for NO reduction by CO on CaO (100) surface in the carbonation stage of CaL process
反应过程各阶段分别为:① CO2分子率先吸附在活性位点上,与CaO(100)表面的O 原子结合形成稳定的 CO结构(R);② NO 分子的N—O 键发生断裂,游离的O 原子与CO 分子结合生成CO2分子,随后吸附在CaO(100)表面(R→IM1→IM2);③ 游离的N 原子与新引入的NO 分子相互靠近逐渐形成IM3, 随后与N—O 键断裂生成的N 原子克服2.91 eV 的反应能垒形成N2分子(IM3→IM4);④ 游离的CO 分子与IM3→IM4 中脱离的O 原子结合,经过TS4 形成1 个CO2分子,最终吸附在CaO(100)表面(IM5→P)。
反应机理中,其中的一个基元步骤进行的速度远较其他速度慢,则该步骤为决速步骤,体现在DFT 计算中就是反应过程中反应能垒最大的一步。由表3碳酸化阶段CaO(100)表面CO 还原NO 各阶段的反应能垒可知,反应过程中的R→IM1→IM2 能垒最高,这是整个反应的决速步骤。
3 结 论
(1) CO 和NO 分子在CaO(100)表面的最佳吸附位是O-top 位,吸附能分别为-0.35 和-0.79 eV,为弱化学吸附。
(2)当CO2、CO 和NO 分子在CaO(100)表面共吸 附 时,Eco-ad,NO比Ead,NO低 约75%,而Eco-ad,CO与Ead,CO相似,这表明在共吸附过程中,CO2与NO 竞争CaO(100)表面的O-top 位,从而抑制了NO 在表面的吸附。
(3)钙循环碳酸化阶段CaO 催化CO 还原NO 的反应过程包括4 个过渡态和5 个中间体,最终生成3个C O离子和1 个N2分子,反应的总能垒为11.08 eV。CO2的形成及在CaO(100)表面的吸附(R→IM1→IM2)为整个反应过程的决速步骤。
