基于苝酰亚胺超分子的新型光自芬顿体系高效降解苯酚的效能与机理
2023-12-11郭英杰李冰爽
郭英杰 , 崔 杨 , 李冰爽 , 李 莹 , 刘 迪
(中国矿业大学(北京) 化学与环境工程学院, 北京 100083)
由于煤炭在我国能源储备中的首要地位,煤化工能源产业发展迅速。同时,煤化工行业也是高耗水行业,煤化工废水中含有各种复杂的污染物,并且污染物的浓度较高,其主要有机污染物为苯酚等酚类物质[1]。在工业净化污水的过程中酚类物质很难被完全除去,经酚回收及物理法/生化法处理后残留的微量苯酚仍会对人体健康产生危害。所以,苯酚废水的高效稳定处理仍是一项难题[2-3]。
以芬顿反应为代表的高级氧化工艺(AOPs),与其他有机废水处理技术相比,具有环境友好、设备简单、反应速度快等优点,其主要通过产生活性氧物种(ROSs)来降解污染物。然而,由于化学氧化剂成本较高,以及与化学生产相关的能源消耗和化学污染,使得AOPs 的发展受到限制[4-5]。此外,AOPs 所需的强氧化性物质(如过氧化氢)的储运存在一定的安全风险[6],且这些氧化剂即使在催化剂存在下产生ROSs的速率仍很低[7],同时其自身也会迅速被消耗,不能保持长期的活性[8]。通过光催化技术原位生成和利用过氧化氢而构建光自芬顿体系,不仅可避免外加过氧化氢而降低成本,也避免了过氧化氢在储运过程中潜在的安全风险,并且结合光催化过程,协同促进ROSs 的生成,进而可实现对污染物的高效降解。同时,光催化氧化技术已被证明在降解苯酚方面具有优异的能力[9]。为了实现光催化处理苯酚废水的工业化应用,选择生产成本低、结构可控性高及元素供给丰富的有机半导体作为可见光驱动型的光催化材料势在必行。
苝酰亚胺(PDI)超分子材料是近几年来备受关注的有机半导体光催化材料之一。PDI 超分子的自组装过程依赖于非共价键相互作用,包括氢键、偶极–偶极相互作用、π-π 堆积作用、疏水–疏水相互作用和静电相互作用等[10]。单分子态的PDI 具有分立且不连续的HOMO 与LUMO 能级,而形成PDI 超分子后,基于小分子间的轨道重叠使得HOMOs 与LUMOs 发生分子间转移积分,进而使PDI 超分子呈现类似无机半导体的带状电子能级结构,并可独立完成从光吸收、载流子分离到催化底物反应的整个光催化过程[11]。PDI 超分子目前已成功应用于光催化领域,包括光催化降解污染物[12-14]及光催化分解水产氢[15]/产氧[16]等。
芬顿反应指过氧化氢(H2O2)和亚铁离子(Fe2+)的混合溶液可将许多已知的有机物质(如羧酸、醇、酯等)降解矿化为无机物(如CO2、H2O 等)[17]。YUE等[18]成功制备了Fe2O3/PDI 复合光催化剂,并用于可见光下催化协同芬顿作用进行苯酚的降解,其降解速率常数分别为Fe2O3和PDI 的4.3 倍和5.5 倍。ZHANG等[19]利用间苯二酚甲醛(RF)高聚物,构建了树脂基光自芬顿体系,可高效地降解和矿化双酚A。JIAN 等[20]围绕着石墨碳环掺杂的石墨相氮化碳(Cg-C3N4)光催化剂,构建出光自芬顿体系,用于高效降解和矿化4-氯苯酚。XIONG 等[21]开发了新型g-C3N4/PDI/Fe 复合光催化剂,并将氮三乙酸(NTA)与Fe3+络合,构建光芬顿体系,大大促进了整体的催化性能。同时有研究表明[22-23],Fe3+可被引入到poly(TA)超分子聚合物网络中,通过与羧基形成强络合位点而取代羧基间的弱氢键作用。此外,PDI 基光催化材料在酸性环境中可原位生成一定量的过氧化氢[24-25]。基于此,为了高效、低成本处理苯酚废水,笔者采用带羧酸侧链的苝酰亚胺超分子(PDI-C)与Fe3+结合,构建了一种新型且高效的PDI-C/Fe3+光自芬顿体系,并对其协同降解苯酚的催化作用机理进行系统研究。
1 实 验
1.1 光催化自芬顿系统
根据文献[26]的方法制备PDI-C 超分子纳米棒,如图1 所示。以咪唑作为溶剂,3,4,9,10-四羧酸酐(PTCDA)与β-氨基丙酸在氩气氛围下110 ℃搅拌反应4 h 后,通过pH 诱导实现超分子的自组装过程,得到超分子产品——PDI-C 超分子纳米棒。在光催化降解苯酚过程中,原位构建出光催化自芬顿体系。其中Fe3+以FeCl3·6H2O 的形式与PDI-C 一同加入到5 mg/L苯酚溶液中,暗吸附1 h 后进行连续的光催化自芬顿体系降解苯酚反应。
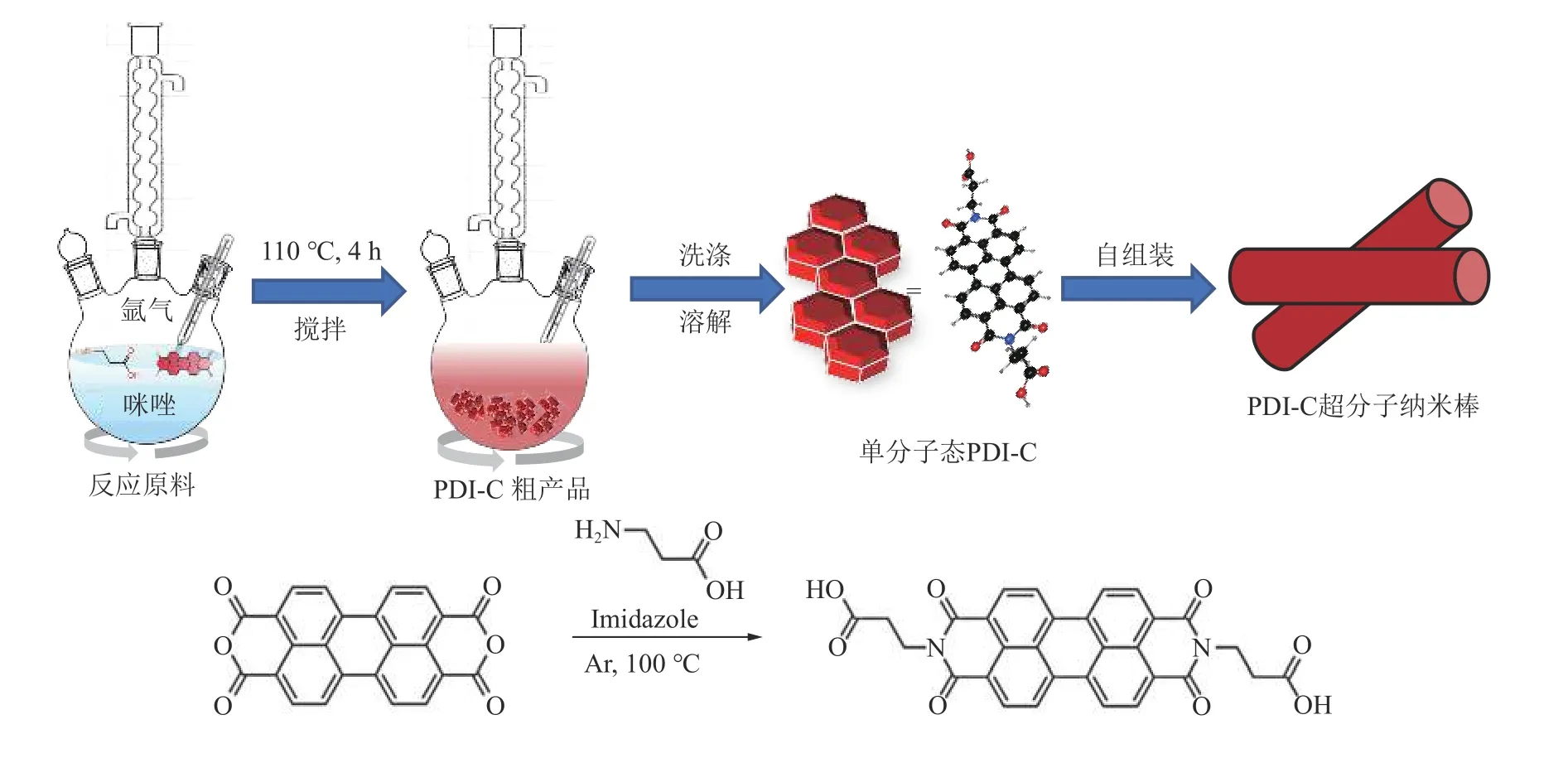
图1 羧酸侧链型PDI 及其超分子的合成路线Fig.1 Synthesis route of PDI with carboxylic acid side chain (PDI-C) and its self-assembly process
1.2 试剂及测试方法
(1) 试剂。3,4,9,10-四羧酸酐、β-氨基丙酸、咪唑、三乙胺、氩气、邻菲罗啉、氯化亚铁四水合物、三氯化铁六水合物、盐酸、氢氧化钠、硝基四氮唑蓝、苯酚、无水乙醇、甲基苯基亚砜、碳酸氢钠、磷酸二氢钠、硫酸钠、硝酸钠、氯化钠。
(2) 测试方法。采用扫描电子显微镜(SEM,HT7700,日本Hitachi 公司)、透射电子显微镜(TEM,SU-8010,日本Hitachi 公司)、高分辨透射电子显微镜(HRTEM,JEM-2100F,日本电子JEOL)观察样品的形貌;采用X 射线衍射仪(XRD, Empyrean, 荷兰帕纳科公司)分析样品的晶体结构;采用傅里叶变换红外光谱仪(FTIR,Nicolet iS20, 德国Bruker 公司)表征样品表面官能团的种类及变化;采用X 射线光电子谱仪(XPS,K-Alpha ,美国赛默飞公司)检测样品的表面元素及其化学状态;采用电子自旋共振仪(ESR,JESFA200,德国Bruker 公司)检测活性自由基种类。
1.3 实验方法
主要采用PDI-C/Fe3+光自芬顿体系催化降解苯酚,并优化了Fe3+添加量、反应体系pH 等降解反应条件;通过追踪催化反应中铁物种形态与含量的变化以及各活性物种的变化,深入探究了可见光催化剂PDI-C协同芬顿作用降解苯酚的过程机理。
其中铁离子浓度采用文献[27]优化的邻菲罗啉分光光度法,Fe4+采用甲基苯基亚砜(PMSO)检验法[28],超氧自由基(·)的定量测量采用硝基四氮唑蓝(NBT)法[29-30]。笔者共进行4 轮循环实验。本体系展现出超高效的第1 轮催化降解率和可观且稳定的后续多轮降解活性(第3、4 轮催化降解率与第2 轮相当)。据此,选取第1、2 轮催化过程作为代表进行催化过程机理的系统探究。
2 结果与分析
2.1 光催化对苯酚降解的影响因素
2.1.1 Fe3+添加量对光催化活性的影响
将25 mg PDI-C 催化剂加入到50 mL 质量浓度为5 mg/L 的苯酚溶液中,并分别投加不同用量的FeCl3·6H2O,在可见光(λ> 420 nm)辐照下进行光催化降解苯酚反应测试。图2(a)为光照10 min 时FeCl3·6H2O 添加量对苯酚降解率的影响。少量(0.1 g/L FeCl3·6H2O)Fe3+的引入即对苯酚的降解产生极大的促进作用。随着Fe3+添加量的增加,苯酚降解率逐步提升,直至添加量为0.4 g/L 时,苯酚的降解率达到最优。综合考虑降解率和经济性,确定Fe3+的最佳添加量为0.4 g/L FeCl3·6H2O,其降解效果为可见光(λ> 420 nm)辐照下可于14 min 内实现对5 mg/L苯酚溶液的完全降解,如图2(b)所示。
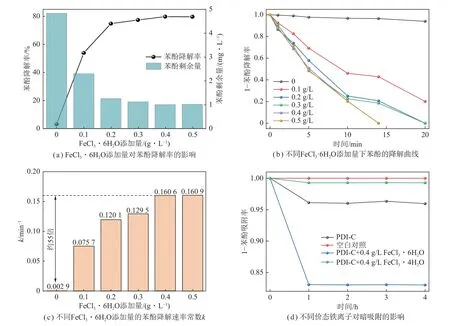
图2 Fe3+添加量对光催化活性的影响Fig.2 Effect of Fe3+ addition on the photocatalytic activity
通过准一级动力学模型对苯酚降解效果进行评价,其降解速率常数相较纯PDI-C 催化剂的体系提升了约55 倍(图2(c)),说明此极高效的PDI-C/Fe3+复合体系的催化降解机理显著区别于常规光催化作用。
此外,笔者还探究了无光照下铁离子与底物苯酚分子(低浓度溶液)间有无明显的相互作用(图2(d)),经与空白样及纯PDI-C(减少约4%)对比发现,加入0.4 g/L FeCl3·6H2O 后苯酚量降低最明显,而添加0.4 g/L 的FeCl2·4H2O 几乎对苯酚底物无影响。因此,暗吸附下苯酚量的明显减少(约15%)应归因于催化剂对苯酚的吸附及Fe3+与苯酚之间的络合作用,使得游离于溶液中的苯酚分子含量相应减少。催化剂对苯酚的吸附与络合作用对苯酚去除率的贡献远小于暗吸附后可见光辐照下对苯酚的催化降解率,因此可排除其对该高效催化反应的主要影响。此外,无论是添加Fe3+还是Fe2+,其在无光条件下吸附–解吸的平衡时间均为1 h,因此,本文所有相关的光催化降解苯酚实验,在光辐照前均取1 h 为暗吸附–脱附平衡的时间。
2.1.2 体系pH 对光催化活性的影响
第1 轮催化反应初始pH=2.55,第2 轮催化反应初始pH=1.58,在pH < 3 时,Fe3+可稳定地以离子形式存在[31]。图3(a)、(b)为第1、2 轮催化反应中体系pH 对降解效果的影响。第1 轮催化反应中,随着盐酸添加量的增加,苯酚的降解效果明显下降。pH=2.55~1.35 时(添加0 ~0.06 mol/L 盐酸),pH 的降低对苯酚降解起抑制作用,因此,第1 轮催化反应中,不添加盐酸,即pH=2.55 时降解效果更优。此外,本文所采用的主体光催化剂是羧酸侧链型苝酰亚胺超分子(PDI-C),相关文献表明[32],碱性条件会影响PDI-C超分子纳米棒的稳定性,因此碱性环境下探究此PDIC/Fe3+体系的苯酚降解活性测试不具备条件。
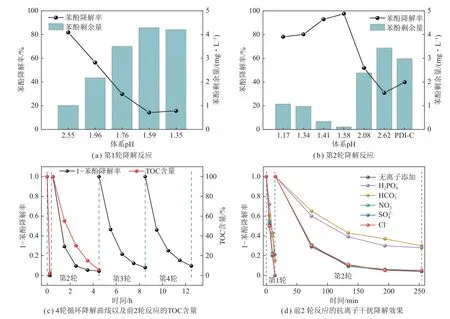
图3 不同反应条件下的光催化降解效果Fig.3 Photocatalytic degradation efficiencies under different reaction conditions.
在第1 轮催化反应中不加入任何酸,继续进行第2 轮催化反应,并探究体系pH 对第2 轮苯酚降解效果的影响,如图3(b)所示。在第2 轮催化反应中,苯酚的降解率相对第1 轮大幅降低,甚至低于同样条件下纯PDI-C 对苯酚的第2 轮降解率,说明仅添加Fe3+在第2 轮催化反应中有可能抑制了苯酚的降解,这类似于催化剂的失活。而在第2 轮催化反应中同时加入一定量的酸时,则可较大幅度提高苯酚降解率。当pH=2.62~1.58(添加0~0.04 mol/L 盐酸)时,随着pH的降低,苯酚的降解效果逐渐提高;而当pH=1.41~1.17(添加0.06~0.1 mol/L 盐酸)时,苯酚的降解效果变差,但仍高于不添加盐酸时。基于此,第2轮催化反应中,盐酸的最佳添加量设定为0.04 mol/L,此时体系pH=1.58。
2.1.3 循环稳定性和总有机碳含量
以 第1 轮 催 化 反 应(25 mg PDI-C, 0.4 g/L FeCl3·6H2O)和第2 轮催化反应(第1 轮中回收得到的PDI-C,0.4 g/L FeCl3·6H2O,0.04 mol/L HCl)为最佳反应条件,在第2 轮催化反应后继续进行循环催化测试,结果如图3(c)所示,第1 轮对苯酚的完全降解需要14 min,第2~4 轮反应则在可见光辐照4 h 后的降解率均超过90%。这与纯PDI-C 超分子的4 轮循环降解率相比提升显著,表明PDI-C/Fe3+体系既显示出高效性,又具备可观的循环稳定性,实际应用潜力大。
为了探究体系对苯酚的矿化能力,对反应过程中总有机碳(TOC)含量变化进行监测。在PDI-C/Fe3+体系中,第 1、2 轮催化反应对苯酚的矿化率均很高,苯酚均可被矿化为CO2和水,此高矿化活性表明PDIC/Fe3+体系具备高效、无二次污染产生的有机污染物处理能力。
2.1.4 抗离子干扰能力
实际水体中广泛存在多种阴离子、阳离子和天然有机物,会影响底物苯酚在催化剂表面的吸附和光自芬顿的降解率。为探究各种阴离子对体系催化效果的影响,在PDI-C/Fe3+体系中分别加入10 mmol/L 的Cl-、S O、N、H C和H2P,结果如图3(d)所示。
2.2 反应过程机理
2.2.1 吸附机理
从苯酚水溶液中吸附底物苯酚分子是PDI-C/Fe3+体系进行光催化反应的首要步骤,对苯酚分子的吸附依赖于催化剂本身的微观结构,但主要受催化剂PDIC 表面性质的影响。PDI-C 对苯酚的吸附可能是由多种相互作用共同控制[35-36]。
使用盐酸将体系pH 调节到2.55(PDI-C/Fe3+体系第1 轮催化反应的初始pH),测量不同体系的Zeta 电位(表1)(Zeta 电位是检测催化剂表面所带电荷数量的物理参数)。由表1 可以看出,PDI-C 在加入Fe2+后,Zeta 电位变得更正,而加入Fe3+后,Zeta 电位由负值变为正值,说明铁离子与光催化剂PDI-C 表面存在一定的相互作用。

表1 不同体系催化剂的Zeta 电位(pH=2.55)Table 1 Zeta potential of various systems (pH=2.55)
需要指出的是,pH 作为影响吸附性能的重要因素之一,对研究吸附剂和吸附质之间静电作用非常重要。通常,pH 既能影响吸附剂的表面电性,又能影响吸附质的存在形式。相关文献表明[36],pH=2.55 时,苯酚主要以分子形态存在于溶液中。
催化剂PDI-C 表面Zeta 电位为负值,说明其表面为负电性,这与其羧酸侧链型分子结构特征相吻合(在水相中部分呈—COO-),其通过静电吸引作用可吸附或络合Fe2+和Fe3+,这是PDI-C/Fe3+和PDI-C/Fe2+Zeta 电位变化的主要原因。
结合2.1.1 节,PDI-C/Fe3+体系暗态下苯酚含量降低:一部分由Fe3+与苯酚分子的络合作用所致,另一部分来源于PDI-C 超分子纳米棒对苯酚的吸附。
2.2.2 表面沉积铁物种成分及其影响
样品形貌与尺寸如图4 所示。PDI-C 自组装形成超分子后呈均一的棒状结构,其直径为20~50 nm,长度为0.8~1.2 μm,长径比超过40。经过第1 轮催化反应后,PDI-C 超分子纳米棒的形貌保持相对稳定,但在棒状结构上发现有明显的附着物(图4(b)、(c)),推测表面形成了铁氧化物的颗粒,这可能与其催化活性下降有关。

图4 反应前后的PDI-C 电镜图Fig.4 Electron micrographs of PDI-C before and after the catalytic reaction
采用高分辨透射电镜对PDI-C 超分子纳米棒上的附着物进行分析,发现其呈清晰的晶格条纹。从图4(d)可以看出,PDI-C 的元素种类在原有的C、O及N 的基础上增加了Fe 元素。结合元素分布和晶面间距测试结果,可以确认表面生成的铁系物为α-FeOOH,其中,晶面间距0.254 nm,可归属为其(111)晶面间的距离,这与ZHONG 等[37]的研究结果一致。
为进一步验证第1 轮催化反应后PDI-C 表面颗粒附着物的定性结果,对其进行了X 射线光电子能谱(XPS)测试。由图5(a)可知,纯PDI-C 中显示3 个峰,分别对应于C=O、C—O 和吸附氧,位于530.9、532.1 和533.4 eV[38-40],而在第1 轮催化反应后这3 个峰仍存在,说明PDI-C 结构在第1 轮催化反应后保持稳定。值得注意的是,在第1 轮催化反应后,出现了一个新的峰(529.7 eV),此峰可归属为α-FeOOH 中的Fe—O 键[37],进一步印证了α-FeOOH 的生成。由图5(b)可知,710.4 eV 和724.0 eV 归属于2p3/2和2p1/2峰,与文献中报道的α-FeOOH[41]相比负向偏移了0.8 eV 和1.1 eV,结合能的降低表明铁的电子密度增加[42],说明铁价态的降低,表明附着物颗粒不是单一的α-FeOOH,还有Fe(II)物种的存在,且由式(1)、(2)可知,附着物中也可能存在Fe(OH)2。将Fe 2p3/2进 一 步 做 分 峰 处 理,Fe 2p3/2的709.6 eV 峰 对 应Fe2+—O, 711.2 eV 峰 对 应 Fe3+—O[43]。 706.4~706.6 eV 处无信号峰,说明表面不存在Fe0[43]。上述测试结果与ZHONG 等[37]的研究结果一致,即反应过程中表面铁氧化物Fe2+—O 和Fe3+—O 不断相互转化(式(1)~(4))。由此可知,表面的铁氧化物在相互转换过程中,会消耗PDI-C 生成的光生空穴(h+),进而使光氧化活性下降,从而解释了表面附着物的存在不利于降解反应。
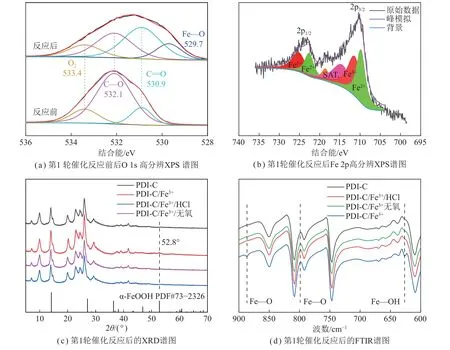
图5 催化反应前后表面铁物种的定性分析Fig.5 Qualitative analysis of surface iron species before and after the catalytic reaction
图5(c)、(d)分别为纯PDI-C、PDI-C/Fe3+经第1轮催化反应后、PDI-C/Fe3+经第1 轮催化反应后酸浸1 h、PDI-C/Fe3+去氧气经第1 轮催化反应后的X 射线衍射(XRD)图谱和傅里叶红外(FTIR)图谱。结果表明,PDI-C/Fe3+第1 轮催化反应后在52.8°出现的较为明显的峰为α-FeOOH 特征峰之一[44]。而PDI-C 和α-FeOOH 在14.1°附近的衍射峰的信号重合,进而可能掩盖了α-FeOOH 的特征峰。纯PDI-C,PDI-C/Fe3+在第1 轮催化反应后的红外图谱如图5(d)所示,其在885 cm–1和790 cm–1处有2 个振动带,对应于α-FeOOH的Fe—O 弯曲振动[44];另外在626.8 cm-1处出现新的振动吸收带,推测为α-FeOOH 中Fe—OH 振动峰红移 所 致[45]。α-FeOOH 的 生 成 可 能 有2 种 路 径:式(1)~(3)和式(5)~(6)。
铁的钝化层也会抑制·OH 的产生,不利于污染物的降解[46]。由此,表面铁物种α-FeOOH 的生成,一方面可能会覆盖PDI-C 超分子纳米棒的表面吸光位点,并阻碍活性物种·OH 的产生;另一方面铁氧化物的生成过程在减少了Fe3+的同时还消耗了体系中的h+等活性物种,进而也抑制了反应的催化活性[31],这可能是第2 轮催化反应在不加酸时苯酚降解率下降的主要原因。为此提出了提高第2 轮催化反应降解率的解决方法:① 尽可能抑制α-FeOOH 的生成。选择在第1 轮催化反应中隔绝氧气,即先煮沸反应用的纯水,并在催化反应中持续通入氩气。用扫描电镜观察第1 轮催化反应后的样品形貌,发现PDI-C 表面几乎观察不到颗粒附着物,如图6(a)所示。② 通过酸浸去除生成的α-FeOOH。将PDI-C/Fe3+第1 轮催化反应后的样品烘干,加入50 mL 的0.04 mol/L 盐酸浸泡1 h(模拟第2 轮催化反应条件)后离心烘干,并通过透射电镜观察其形貌,如图6(b)、(c)所示。与第1 轮反应后样品相比,表面生成的α-FeOOH 被溶解除掉。XRD 和FTIR 的表征结果也证实了这一点,说明适当浓度的酸洗可去除PDI-C 表面生成的α-FeOOH 等铁氧化物层。同时,加酸还可使反应(11)向右进行,促进高活性 ·OH 的产生,进而在一定程度上恢复光催化反应的效率。

图6 不同条件下反应前后的PDI-C 的电镜图Fig.6 Electron micrographs of PDI-C before and after reaction under different conditions
综上,第1 轮催化反应中隔绝氧气或第1 轮催化反应结束后加酸,均可有效避免铁氧化物的沉积。而隔绝氧气的方式在后文中被证明会抑制ROSs 的产生,进而对光催化氧化降解过程起负面作用,因此2~4轮循环催化过程均采取加酸处理。
2.2.3 反应液中铁物种含量变化及其作用
将第1 轮催化反应后的分散液离心保留上清液,补充新鲜的PDI-C 以及5 mg/L 苯酚进行降解,在20 min 内可将5 mg/L 苯酚完全降解,相比于原第1轮降解的14 min 略有下降,这可能是由于铁物种形态或含量发生变化所致。
为了了解反应液中铁物种对降解苯酚发挥的作用,研究了反应过程中溶液中可能存在的铁物种种类:Fe2+、Fe3+、Fe4+。在4 轮循环反应中加入PMSO,每隔1 h 检测反应溶液中PMSO 和PMSO2的浓度变化,如图7(a)所示,PMSO 和PMSO2的浓度始终在小幅波动,说明PMSO 未转化成PMSO2,即4 轮循环过程中均不存在Fe4+[47]。不同循环阶段结束时溶液中Fe2+和Fe3+的浓度变化如图7(b)所示。第1 轮循环后Fe3+大幅被消耗,显示了其作为电子受体的作用[42,48]。同一轮循环结束时,溶液中Fe2+浓度大幅上升,且有36.125%的Fe3+转化为Fe2+。Fe3+是常见的电子捕获剂[48],即Fe3+和光生电子结合生成Fe2+,一方面可促进光生电子和空穴的分离,进而促使光生空穴发挥高效光氧化作用;另一方面Fe2+结合光催化原位生成的H2O2可发挥出高效的芬顿催化作用(光自芬顿),这是在第1 轮反应中PDI-C/Fe3+体系具有优异的苯酚降解率的原因。而第2 轮循环结束时,溶液中Fe2+浓度大幅下降,同时Fe3+浓度升高,Fe3+和Fe2+浓度比上升;第3 轮循环结束时,Fe2+和Fe3+浓度均下降,但Fe3+和Fe2+浓度比相对于第2 轮结束时仍为升高趋势。传统芬顿反应中,Fe2+以式(11)的方式被氧化为Fe3+,其反应速度较快,而式(12)的Fe3+还原速度很慢,即铁离子的还原速率远低于氧化速率,较差的铁离子对循环限制了芬顿反应高效、持久的运转[49]。而光催化与芬顿协同作用,则可通过光生电子还原Fe3+的方式使铁离子对的循环变得高效持久,因而产生了“1+1 > 2”的促进效果。
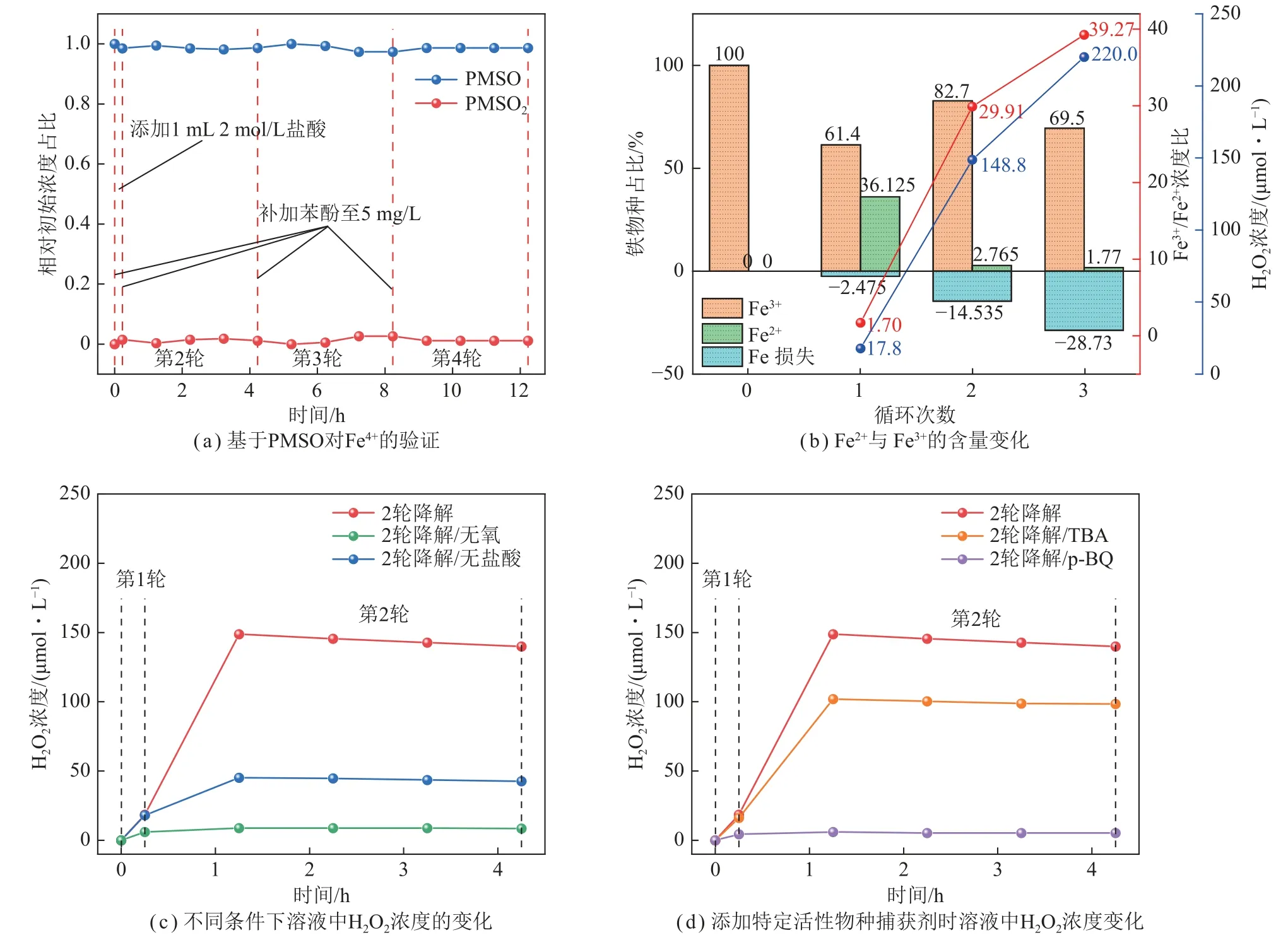
图7 反应液中铁物种和H2O2 浓度变化Fig.7 Changes in the concentration of iron species and H2O2 in the reaction solution
2.2.4 双氧水浓度的变化及其产生机制
光催化诱导双氧水的产生一般有2 种路径[50]:① 光生空穴氧化水,即水氧化反应(WOR),如式(7)、(8)所示;② 光生电子还原氧气,即氧气还原反应(ORR),如式(9)、(10)所示。在降解过程中,第2 轮催化反应进行到1 h 后,断开光源,让其在黑暗环境下静置24 h,过滤取其上清液,检测此上清液中的苯酚含量和总有机碳含量,发现总有机碳和苯酚含量几乎均为0。由此说明PDI-C/Fe3+体系生成了某种新的氧活性物种,推测为芬顿作用中的重要反应物——双氧水,使得体系继续在无光照下发挥了降解作用。
第1、2 轮反应过程中双氧水浓度随反应时间的变化如图7(c)、(d)所示。在第1 轮催化反应结束时,双氧水浓度为17.8 μmol/L,而未加Fe3+的第1 轮催化反应结束时产生的双氧水浓度为3.2 μmol/L,可见Fe3+的加入使同条件下双氧水浓度提升了4.5 倍。第2 轮催化反应光照1 h 后,双氧水浓度大幅增加,达到148.8 μmol/L,之后则轻微下降,4 h 后双氧水浓度为139.9 μmol/L,可见第2 轮催化反应过程中双氧水浓度水平较高且保持相对稳定。原位生成的双氧水可与铁离子形成芬顿体系,促进了苯酚的降解,这也是PDI-C/Fe3+体系4 轮循环下高效降解苯酚的主要原因。第2 轮光催化反应1 h 后停止光照,反应液静置24 h后实现了对苯酚的矿化,进一步证实了芬顿反应的存在。需要指出的是,据文献[51-53],双氧水浓度过高时,并不利于光芬顿反应,一方面过氧化氢可分解产生氧气和水,另一方面过高的双氧水也会成为部分自由基活性物种的俘获剂,这也是第2~4 轮反应降解速率低于第1 轮的原因。
通过探究光催化自产双氧水的影响因素,可确定双氧水产生的主要途径。不同条件下双氧水浓度的变化曲线如图7(c)所示。第2 轮催化反应表明酸的加入促进了双氧水的生成,可能是由于加酸促进了反应式(10)的进行。在催化反应中隔绝氧气,双氧水浓度均远低于有氧时,说明溶解的氧气是光催化自产双氧水的重要原料,且可确定路径②为自产双氧水的主要途径。同时,隔绝氧气下仍有较低浓度双氧水的产生,说明路径①应作为次要途径也存在。为进一步探明本体系光催化自产双氧水的2 种途径及其主次要关系,采用加入活性物种捕获剂的方式研究不同活性物种对光催化诱导双氧水产生的影响,如图7(d)所示。加入·OH 捕获剂叔丁醇(TBA)时,第1 轮反应对双氧水的产生几乎无影响,说明第1 轮催化反应中双氧水的产生不由路径①决定;而双氧水在第2 轮催化反应后浓度下降较为明显,降为101.8 μmo/L,说明此时路径①对双氧水生成的贡献增大。加入 ·捕获剂对苯醌(p-BQ)时,第1 轮催化反应结束时双氧水浓度降至4.6 μmol/L,在第2 轮催化反应后,浓度下降为5.7 μmol/L,说明路径②(2e–ORR)是双氧水主要的生成方式。
2.2.3 节中铁损失随着4 轮循环反应的进行而持续增加,此铁损失来源于铁氧化物种的产生(α-FeOOH、Fe(OH)2),因此,铁损失、Fe2+占比降低(因Fe2+可通过式(11)分解H2O2以产生高活性的·OH)及较高的H2O2浓度是后续循环过程降解率相对第1 轮反应明显降低的原因。
2.2.5 反应过程活性物种的定性与定量
为了检测PDI-C/Fe3+光催化反应过程中的活性物种,采用电子顺磁共振(ESR)进行表征。选用5,5–二甲基–1–吡咯啉–N–氧 化物(DMPO)作为·OH 和·的捕获剂,选用2,2,6,6–四甲基哌啶氧化物(TEMPO)作为单线态氧(1O2)的捕获剂,选用5,5–二甲基–1–吡咯啉–N–氧化物(CPH)作为空穴(h+)的捕获剂,结果如图8 所示。
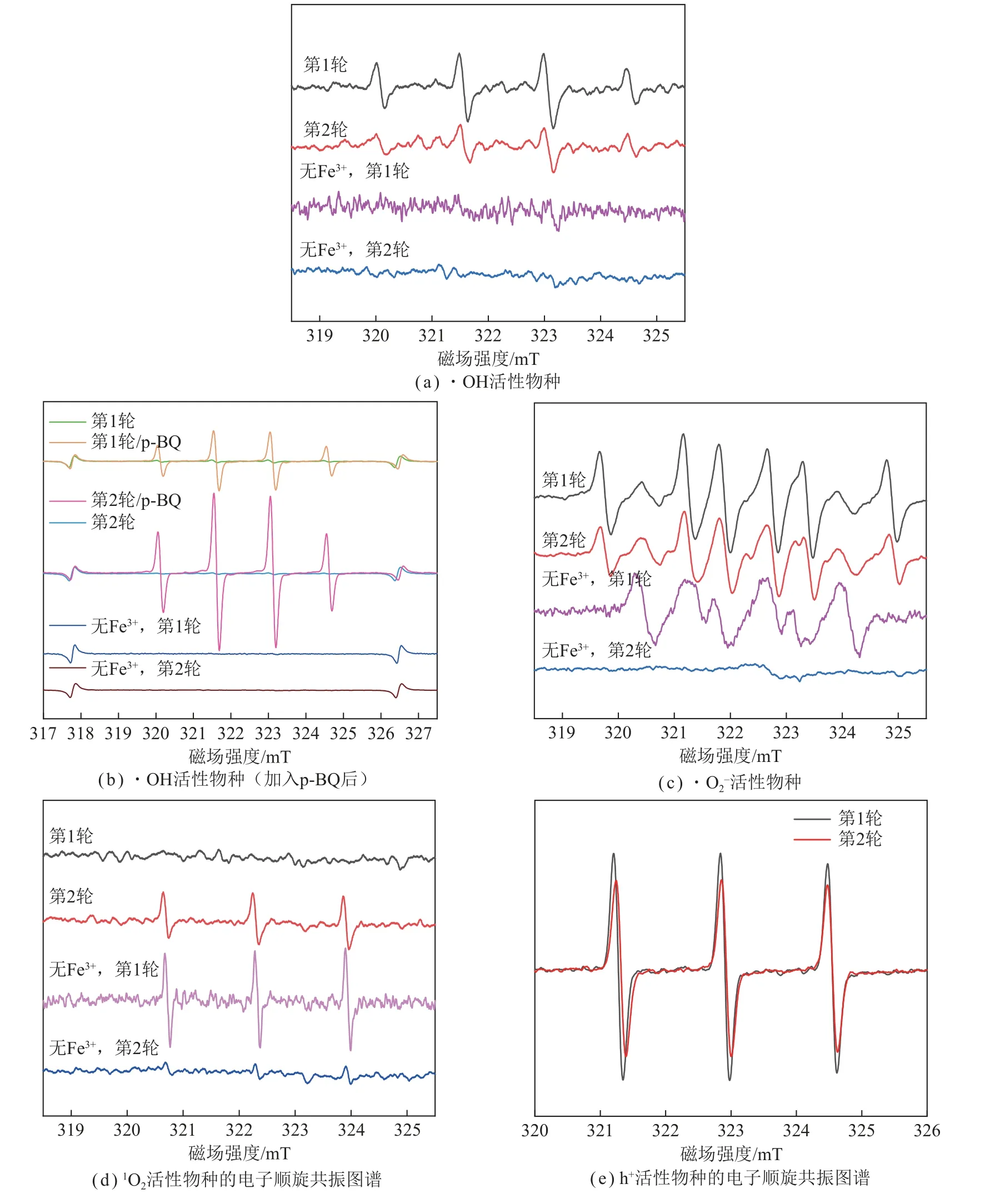
图8 反应过程中活性物种的检测Fig.8 Detection of active species in reaction processes
第1、2 轮降解过程中有明显的·OH 信号峰(图8(a)),说明PDI-C/Fe3+体系可在可见光下产生·OH。而未加Fe3+的降解过程中,未发现明显的·OH 信号峰,这与WANG 等[26]的研究结果一致,说明Fe3+对PDI-C 在可见光下产生·OH 起关键作用。同时,WANG 等[54]证明了H-PDI(同本文PDI-C)和JPDI 的价带顶位置均比H2O/·OH 氧化电位更负,即热力学上其无法实现氧化水产生·OH。而本研究中,通过能斯特方程计算,第1 轮反应pH=2.55 时H2O/·OH 的理论氧化还原电位为2.01 V,第2 轮反应pH=1.58 时H2O/·OH 的理论氧化还原电位为1.95 V,均高于PDI-C 的氧化电位1.58 V,说明PDI-C在这2 种pH 下无法产生·OH,所以体系中的·OH均来源于芬顿反应过程(式(11))。如2.2.2 节所述,第2 轮催化过程中加酸不仅可促进式(10)中双氧水的生成,还可与式(11)中产生的OH-发生反应,促进·OH 的高效产生,从而有利于芬顿体系发挥作用。
图8(b)为原位加入特定活性物种捕获剂时ESR对各活性物种的检测结果。在第1、2 轮降解反应中加入 ·捕获剂p-BQ,·OH 信号大幅提高,说明·和·OH 的生成存在竞争关系,即可能通过式(11)、(12)产生竞争。需要指出的是,PDI-C 同样能通过能量转移过程产生1O2。图8(d)为第1、2 轮降解反应中的1O2信号。其中,第1 轮催化反应中未出现明显的1O2信号峰,说明PDI/Fe3+体系在第1 轮催化反应中主要以电子转移机制为主;第2 轮催化反应中出现明显的1O2信号峰,说明PDI-C/Fe3+体系在第2 轮催化反应中同时存在能量转移机制[54]。由图8(e)可得,降解过程中存在h+,且第2 轮反应中h+含量略有下降。
综上,最优条件下第1 轮催化反应中,主要的活性物种为h+、·OH 和 ·,第2 轮催化反应中主要的活性物种有h+、·OH、 ·和1O2。2 轮催化反应中,在铁离子存在下,·OH 和 ·的生成存在竞争。
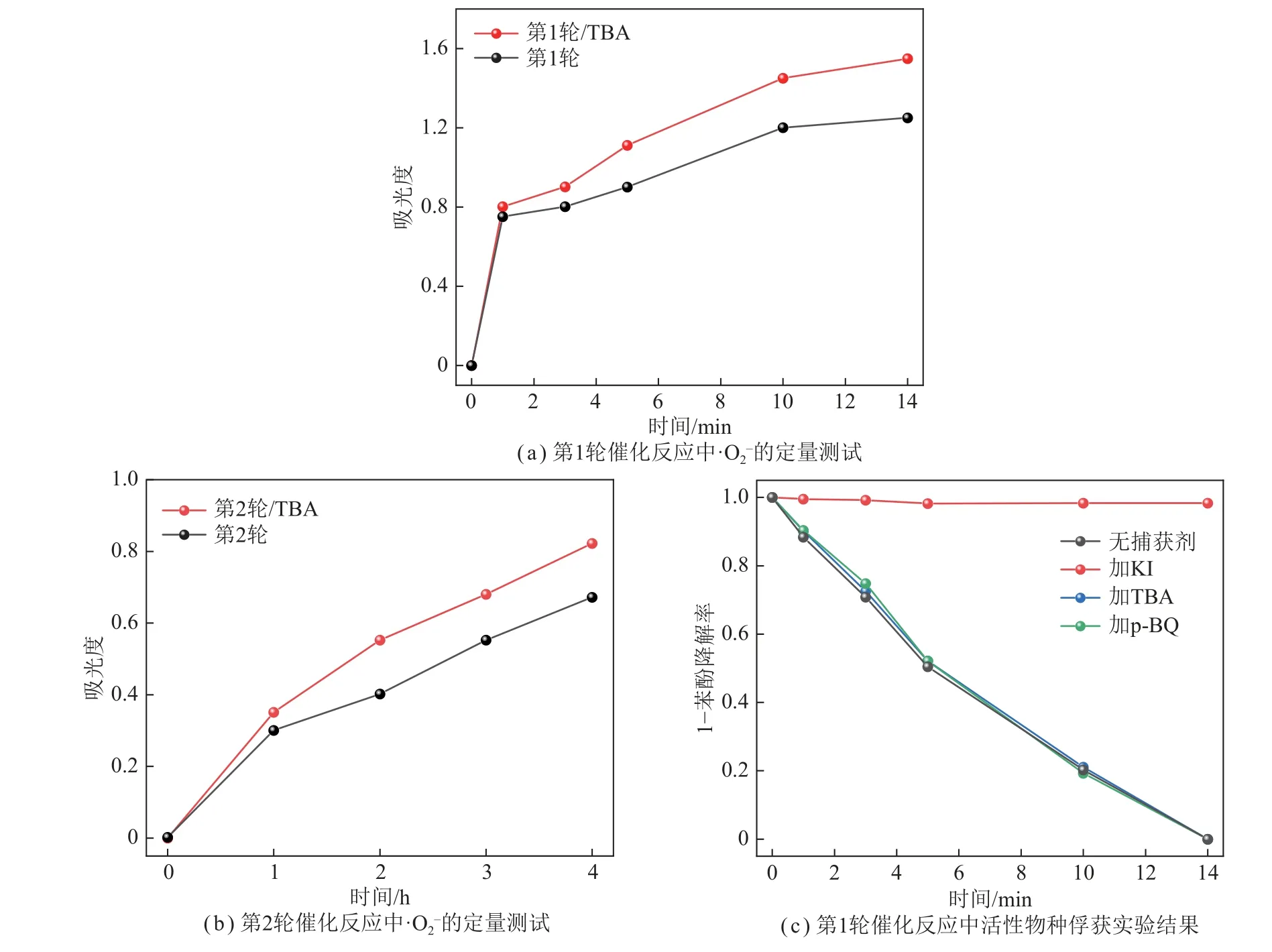
图9 反应过程中活性物种的定量检测Fig.9 Quantitative detection of active species in reaction processes
笔者研究了单一活性物种缺失对PDI-C/Fe3+体系降解苯酚效率的影响,通过向第1、2 轮催化反应中原位添加活性物种捕获剂,得到降解苯酚效果,如图9(c)所示。在第1 轮催化反应中无论是加入·OH还是 ·捕获剂,PDI-C/Fe3+体系苯酚降解率均未发生明显变化,仍为14 min 内高效降解苯酚,这与ESR所揭示的存在h+、·OH 和 ·三种活性物种的实验结果不对应。推测可能是:① 第1 轮催化反应中·OH和 ·的生成之间存在竞争关系,即减少的·OH 或·会使得对应的另一活性物种含量增加,进而导致光催化苯酚降解率不变;② 在第1 轮催化反应中加入h+捕获剂时,PDI-C/Fe3+体系几乎无降解效果,说明h+是第1 轮催化反应的主要活性物种,同时在捕获h+的过程中影响其他活性物种的生成。综上,第1 轮反应中h+是主要的活性物种,电子捕获剂Fe3+的加入促进了光生e–与h+的有效分离,进而实现了对h+的高效利用,而原位产生的双氧水与铁离子对组成的芬顿体系促进了苯酚的降解反应;而在第2~4 轮反应中,高浓度双氧水的生成、铁离子对的有效循环、合适的pH使芬顿体系继续发挥重要作用,进而实现对苯酚降解的高循环稳定性。光催化反应机制模型如图10 所示。

图10 光催化反应机制模型示意Fig.10 Schematic diagram of photocatalytic reaction mechanism
3 结 论
(1) 开发出一种新型的PDI-C/Fe3+复合体系,研究发现Fe3+的存在对PDI-C(羧酸侧链型苝酰亚胺)超分子催化剂可见光下多轮循环降解苯酚的过程发挥了极高的协同促进作用,即构成了高效的光自芬顿系统。此PDI-C/Fe3+光自芬顿系统,作为环境友好且高效的污水处理异相催化剂,可展现出巨大的应用前景。
(2) 通过对催化过程作用机制的系统探究,本研究证实了Fe3+通过与PDI-C 超分子羧酸侧链位点的相互作用,一方面发挥其电子捕获剂的作用,接受光生电子转变为Fe2+,大幅促进了体系光生载流子的分离及铁离子对的循环;另一方面,光催化过程原位生成的双氧水,与铁离子形成了芬顿体系,协同促进了苯酚的高效、稳定降解。
