气相色谱法测定三七粉及含三七原粉类中成药中17种有机氯类农药残留
2023-12-07谢秋红邵大志马晓静宋瑩崔业波马彧
谢秋红,邵大志,马晓静,宋瑩,崔业波,马彧
(1.四平市中心医院,吉林四平 136000; 2.四平市食品药品检验所,吉林四平 136000)
三七为五加科三七的干燥根及根茎,具有散瘀止血,消肿定痛功效。三七粉在临床使用由来已久,尤其在我国北方地区深受患有心血管疾病老人的欢迎,而三七原粉入成药的品种,《中华人民共和国药典》2020 年版一部收载的大约有70 多个品种,在部颁标准中收载的大约有30多个品种,含三七药粉的药品临床疗效确切,利用价值高。近年来三七市场需求量巨大,每年约3.5 万吨[1-6]。中药材种植者施用农药防治病虫害是提高中药材产量的有效措施,但同时也影响了中药材的品质,增大了用药风险。
《中华人民共和国药典》(以下简称《中国药典》)2020年版一部中收载的三七质量标准中,没有有机氯类农药残留量检查项。而GB 2763—2021《食品中农药最大残留限量标准》、《药用植物及制剂外经贸绿色行业标准》、《无公害三七药材及饮片的农药残留与重金属及有害元素残留限量》、《中华人民共和国药典》(2020 年版)“药材和饮片检定通则”对33种禁用农药的残留限量做出规定,WM/T2-2004 则规定了有机氯农药的残留限量[7]。T/CATCM 003—2017 团体标准参考《中国药典》及美国、欧盟、日本和韩国等国家及地区相关标准,对三七中206 种农药制定了限量标准,是我国记载三七农药残留限量最全的标准,但普及度和法律效应不及GB 2763—2021和《中国药典》。
三七药材中有机氯类等农药残留情况报道和研究较多[8-13],且五氯硝基苯超标的情况时有报道,现阶段对中成药中有机氯类农药的测定方法报道较少[16-17],在食品、水果和蔬菜中研究报道较多[18-21],检测方法多为气相色谱法。《中华人民共和国药典》四部通则2341第一法也为气相色谱法,该法存在氧化氯丹与δ-六六六、顺式环氧七氯与反式环氧七氯、p,p'-滴滴滴与p,p'-滴滴涕等组分的分离效果不佳和样品处理繁琐等缺点。笔者在此基础上,采用DM-1701 毛细管柱,优化色谱条件,使17 种有机氯类农药组分均达到基线分离。该方法具有样品处理简单、分析速度快、灵敏度高、专属性好的特点。
1 实验部分
1.1 主要仪器与试剂
气相色谱仪:7890B 型,配63Ni-ECD 电子捕获检测器,美国安捷伦科技有限公司。
电子天平:BP211D型,感量为0.1 mg,北京赛多利斯仪器天平有限公司。旋转蒸发仪:EⅤ311型,莱伯泰科有限公司。
高速离心机:TG16-WS 型,湖南湘仪实验室仪器开发有限公司。
二氯甲烷、丙酮和正己烷:色谱纯,成都市科隆化学品有限公司。
硫酸、无水硫酸钠和氯化钠:分析纯,国药集团化学试剂有限公司。
有机氯农药混合对照品溶液:含有六氯苯、α-六六六、五氯硝基苯、γ-六六六、七氯、艾氏剂、β-六六六、氧化氯丹、δ-六六六、顺式环氧七氯、反式环氧七氯、反式氯丹、顺式氯丹、p,p'-滴滴伊、o,p'-滴滴涕、p,p'-滴滴滴、p,p'-滴滴涕17 种有机氯农药,质量浓度依次为10.03、9.71、9.90、9.62、9.84、9.77、8.89、9.79、7.55、9.73、9.65、9.46、9.32、9.86、9.92、9.32、9.28 μg/mL,批号为610005-202003,中国食品药品检定研究院。
五氯硝基苯对照品溶液:1 000 mg/mL,批号为A1907264,坛墨质检标准物质中心。
三七粉及含有三七成分中成药样品:共23 批,其中三七粉3 批、中成药6 种20 批,包括三七伤药片、丹七片、腰息痛胶囊等,市售,具体信息见表1。
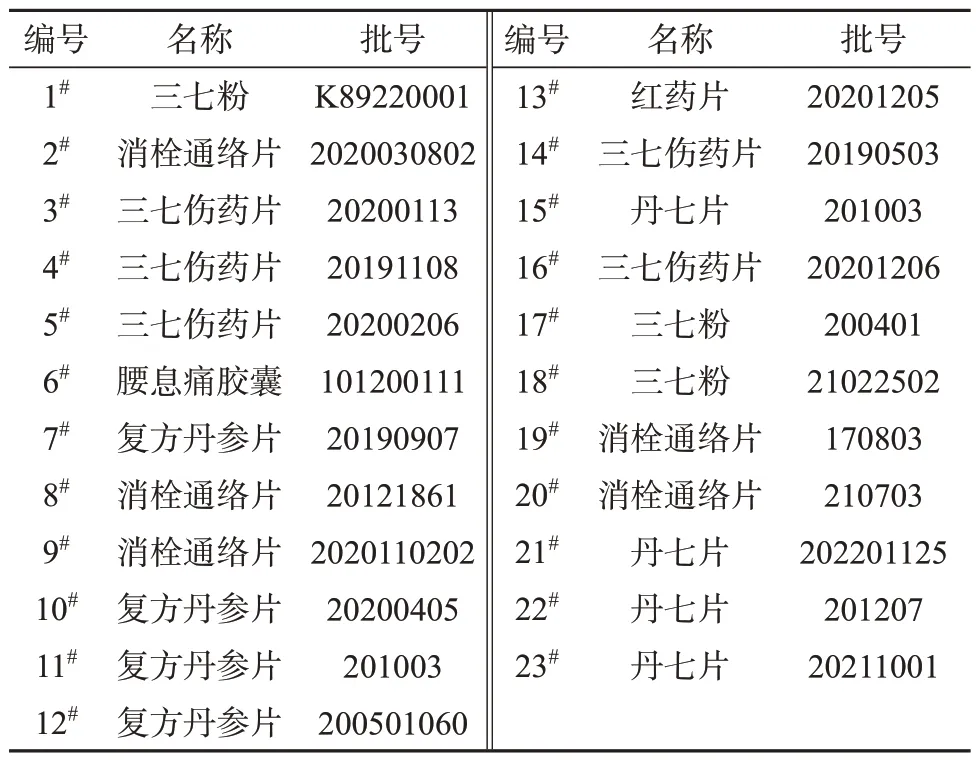
表1 样品信息
1.2 仪器工作条件
分析柱:DM-1701型毛细管柱(30 m×0.32 mm,0.25 μm,北京迪马克科技有限公司);载气:氮气;流量:1.5 mL/min;进样口温度:230 ℃;进样方式:不分流进样;色谱柱温度:程序升温,初始温度为60 ℃,保持0.5 min,以10 ℃/min 的速率升至240 ℃,保持3 min,再以20 ℃/min 的速率升至280 ℃,保持5 min;63Ni-ECD电子捕获检测器温度:300 ℃。
验证柱:DB-5 型毛细管柱(30 m×0.32 mm×0.25 μm,美国安捷伦科技有限公司);载气:氮气;初始流量:1.5 mL/min,恒压模式;色谱柱温度:程序升温,初始温度60 ℃,保持0.3 min,以60 ℃/min 的速率升至170 ℃,再以10 ℃/min 的速率升至220 ℃,保持10 min,再以1 ℃/min的速率升至240 ℃,再以15 ℃/min 的速率甚至280 ℃,保持5 min;其它条件同分析柱。
1.3 溶液配制
有机氯农药混合对照品储备液:0.1 μg/mL。准确移称取有机氯农药混合对照品溶液1 mL,置于100 mL 的容量瓶中,用正己烷定容至标线,混匀,0~4 ℃冰箱中密封保存。
有机氯农药系列混合标准工作溶液:依次移取有机氯农药混合对照品储备液适量,用正己烷稀释配制质量浓度分别为5、10、20、50、100 ng/mL 的有机氯类农药系列混合标准工作溶液,现用现配。
五氯硝基苯标准溶液:0.1 mg/mL。准确移取五氯硝基苯对照品溶液1 mL,置于100 mL 的容量瓶中,用正己烷定容至标线,混匀,再移取1 mL,置于100 mL的容量瓶中,用正己烷定容至标线,混匀,于0~4 ℃冰箱中密封保存。
1.4 样品处理
按日服用量,称取三七粉或含有三七成分中成药样品,置于具塞锥形瓶中,加入水30 mL,振摇使完全崩解(或溶散),准确加入丙酮50 mL,称定质量,超声30 min,放冷,再称定质量,用丙酮补足减失的质量。加入氯化钠约8 g,准确加入二氯甲烷25 mL,称定质量,超声15 min,再称定质量,用二氯甲烷补足减失的质量,振摇使氯化钠充分溶解,静置,转移至离心管中,以3 000 r/min 离心3 min,将有机相转移至装有适量无水硫酸钠的具塞锥形瓶中,放置30 min。准确移取15 mL,于40 ℃减压浓缩至约1 mL,加入正已烷约5 mL,减压浓缩至近干,用正己烷溶解并转移至5 mL容量瓶中,用正己烷定容至标线,摇匀,移至离心管中,缓缓加入硫酸溶液(硫酸与水体积比为9∶1)1 mL,振摇1 min,以3 000 r/min 离心10 min,分取上清液,加水1 mL,振摇,取上清液,作为样品溶液。同法制备空白溶液。
1.5 实验方法
取样品溶液和有机氯农药系列混合标准工作溶液各1 μL,注入气相色谱仪中,按照1.2项下分析柱色谱条件分析,得各农药残留对照品线性方程;样品溶液中如检出与相应农药残留对照品保留时间相一致的色谱峰,则取对应浓度的有机氯农药对照品溶液,连续同法分析3次,取3次平均值,按外标法计算其含量,并用验证柱对检出成分进行验证。
2 结果与讨论
2.1 色谱柱的选择
《中国药典》2020 年版四部通则2341 第一法9种有机氯农药残留测定法[14],以(14%-氰丙基-苯基)甲基聚硅氧烷或(5%苯基)甲基聚硅氧烷为固定液,依法操作,p,p'-滴滴滴与p,p'-滴滴涕无法充分分离。22 种有机氯农药残留测定法,以50%苯基-50%二甲基聚硅氧烷为固定液,依法操作,氧化氯丹与δ-六六六和顺式环氧七氯与反式环氧七氯等多组分均无法充分分离。因此,选择固定液为键合交联14%氰丙基苯基二基硅氧烷的色谱柱进行试验。
分别对同规格30 m×0.32 mm,0.5 μm的色谱柱OⅤ1701 柱、DM1701 柱和DB1701 柱进行筛选,17种有机氯类农药对照品在上述三款色谱柱上的分离效果有明显差异。DM-1701柱分离效果最好,色谱图见图1,空白溶液在16.8 min 时有一色谱峰,与7号色谱峰保留时间17.1 min不一致;而DB1701柱采用恒压模式,初始流量为1.5 mL/min,程序升温同验证柱条件,可较好地分离17个农药对照品。试验结果表明,OⅤ1701柱较难获得较好的色谱图,通过调整程序升温条件、氮气流量及分析模式,β-六六六、氧化氯丹、δ-六六六和顺式环氧七氯的分离效果仍不理想。考虑到样品中可能有杂质对结果有干扰,需要用DB-5 柱进一步验证,笔者对19 批检测出五氯硝基苯的样品采用DB-5柱对分析柱的检测结果进行验证,验证结果均可得到与五氯硝基苯对照品保留时间相一致的色谱峰,证明检测结果无误。综合上述,最后选取DM-1701色谱柱。
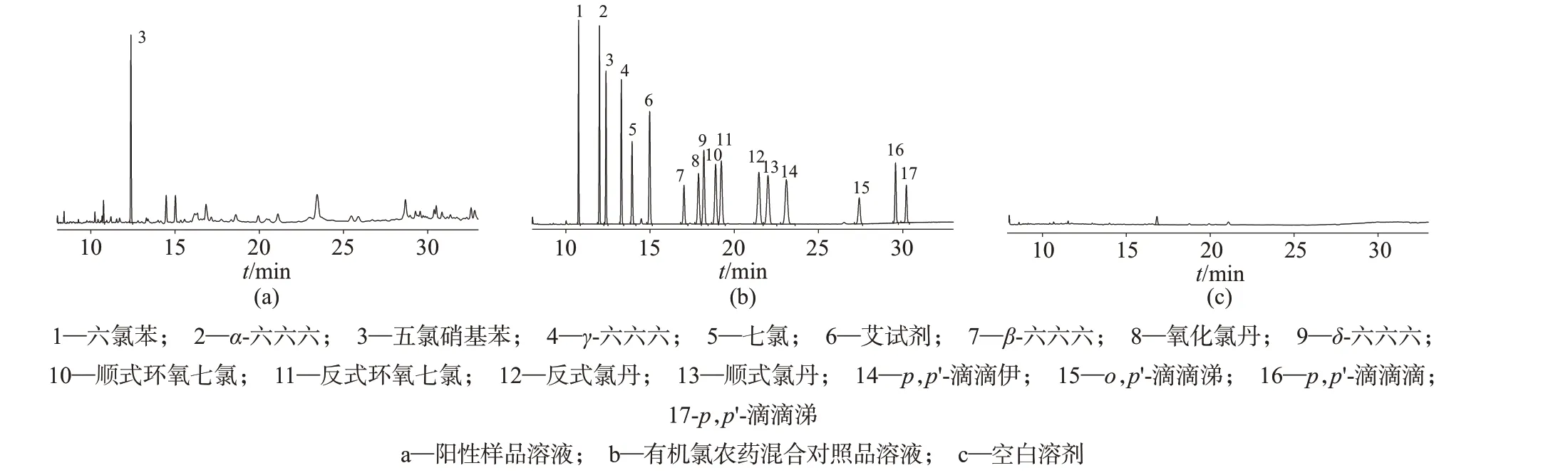
图1 DM1701分析柱气相色谱图
2.2 柱温
《中国药典》2020 年版四部通则2341 第一法分别针对9种和22种有机氯类农药残留分析,均采用程序升温,但依法操作,均难以得到理想的分离效果图。参考人参药材质量标准[15],优化程序升温条件,将原来5个阶段程序升温优化至三个阶段。初始温度保持时间延长至0.5 min,增加各组分的保留值;第二阶段柱温提高至240 ℃,保持3 min,升温速率为10 ℃/min,确保氧化氯丹与δ-六六六和顺式环氧七氯与反式环氧七氯达到基线分离;第三阶段柱温280 ℃,保持5 min,升温速率提高至20 ℃/min,确保o,p'-滴滴涕检出的同时,缩短分析时长。
2.3 样品处理
《中国药典》2020 年版四部通则2341 第一法22种有机氯类农药残留分析方法,样品通过乙腈提取后需要经凝胶渗透色谱柱净化,然后再经过弗罗里硅土固相萃取柱净化,操作繁琐,且准确度不高。取样品日服用量,参考9 种有机氯类农药残留分析方法和人参质量标准,用硫酸对溶液进行净化,置换试剂改为正己烷,消除溶剂干扰的同时,并缩短样品处理时间,提高方法的准确性。
2.4 线性关系和检出限
取有机氯农药系列混合标准工作溶液,按照1.2分析柱色谱条件,分别进样分析,记录色谱图,以各农药质量浓度为横坐标,以对应的色谱峰面积为纵坐标,绘制标准曲线,计算线性方程及相关系数。
取农药残留对照品溶液10 ng/mL,用正己烷适当稀释,按照信噪比法计算各农药残留对照品检出限,以信噪比约为3∶1 时对应的质量浓度作为对应组分的检出限。
17 种有机氯农药线性关系及方法检出限列于表2。由表2数据可知,各组分线性相关系数均不小于0.998,表明在对应浓度范围内线性关系良好。
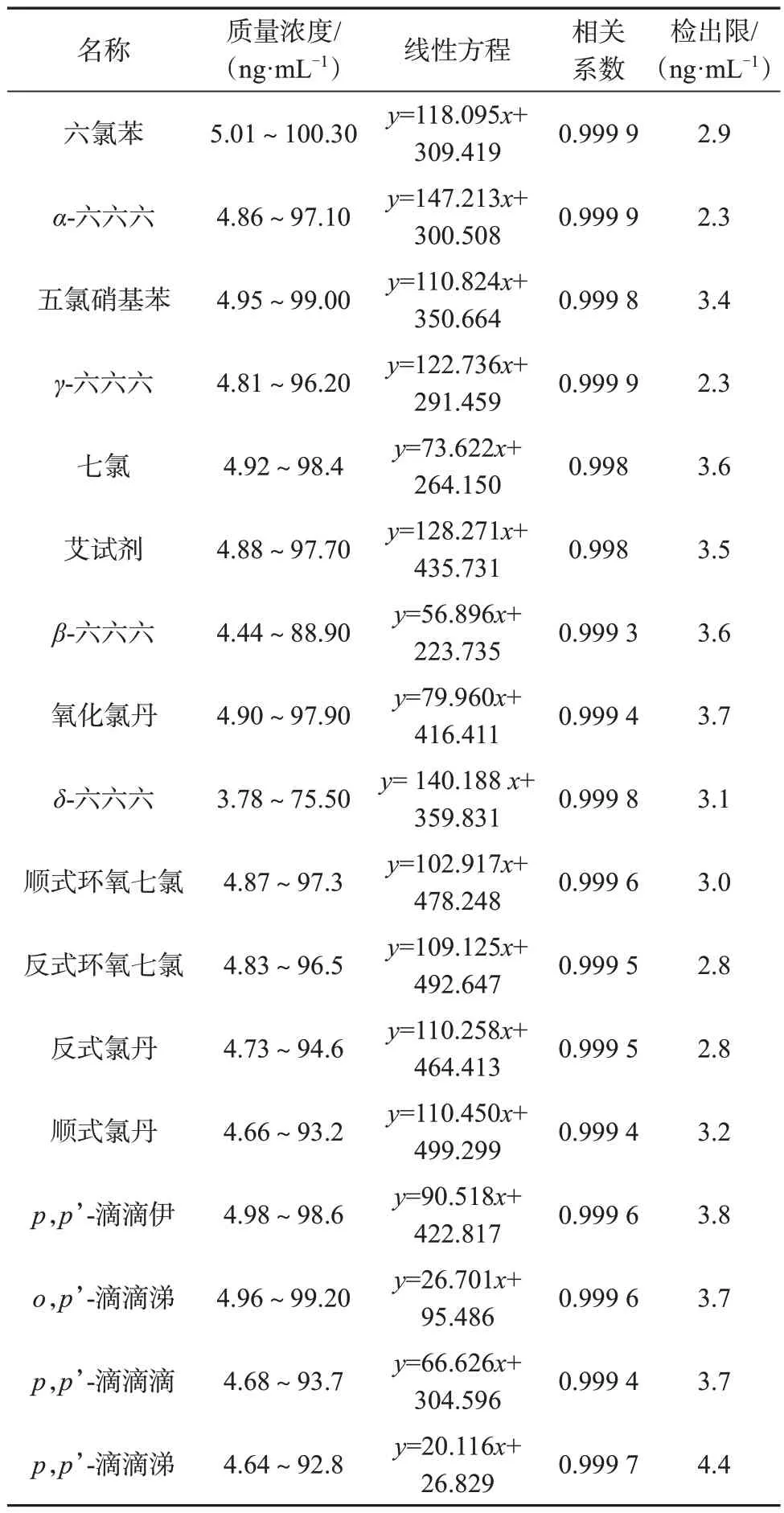
表2 17种有机氯农药线性关系及方法检出限
2.5 样品加标回收试验
取上述23 批次样品,用该方法测定,有19 批样品检出五氯硝基苯,其它农药组分均未检出。19批阳性样品中,丹七片等5批样品超出限度,存在一定的安全隐患。因此,按照丹七片处方工艺,取丹参250 g 按照处方工艺提取浓缩,不添加三七粉,加入等量的淀粉与糊精混匀,制备阴性样品。
取上述阴性样品3.5 g,精密称定,共计6 份,置于具塞三角瓶中,准确量取2 mL有机氯农药混合对照品储备液(0.1 μg/mL),待低温挥干溶剂后,按照1.4方法处理,得加标样品溶液,照1.2分析柱色谱条件分析,以色谱峰面积外标法定量,计算回收率及相对标准偏差,结果列于表3。由表3可知,17种有机氯类农药平均回收率为90.3%~102.2%,相对标准偏差为1.8%~2.8%,表明该方法准确度、精密度均良好。
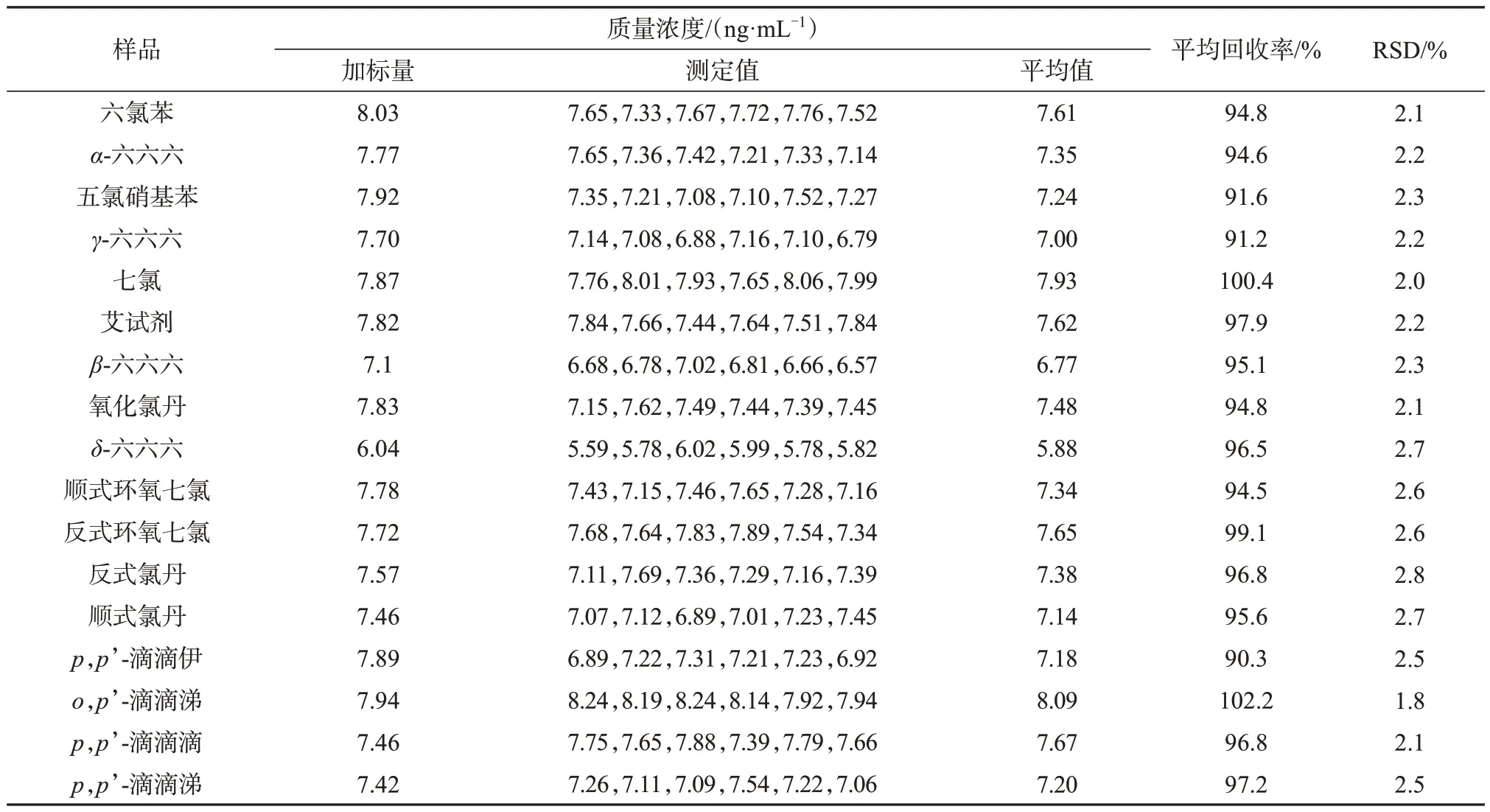
表3 阴性样品加标回收试验结果
3 结语
用改进条件的气相色谱法对丹七片等6种中成药和三七粉中17种有机氯农药残留量进行测定,该方法灵敏度高,分析速度快,准确度和精密度良好。
