限进介质色谱柱高效液相色谱法测定动物源性食品中对乙酰氨基酚残留量
2023-12-07宁霄谢宏洋杨茜崔粲黄圣南
宁霄,谢宏洋,杨茜,崔粲,黄圣南
[1.中国食品药品检定研究院,北京 100050; 2.海口海关技术中心,海口 570311;3.三耀精细化工品销售(北京)有限公司,北京 100022]
限进介质色谱的特点在于可以排除蛋白等大分子的影响直接对目标小分子进行检测,而存在大分子干扰问题的不仅仅存在于生物样品分析检测中,在食品的液相色谱、液相色谱-质谱分析领域同样存在严重的基质干扰问题,特别是动物源性食品分析。在检测过程中,通常为了排除这一类基质的影响,必须选择复杂的样品预处理方法,如液液萃取(LLE)法[8]、固 相 萃 取(SPE)法[9-12]、“QuChERS 原则”[13-14]等,或多种方法组合使用,通常需要耗费较高耗材成本和大量分析时间。
对乙酰氨基酚是临床常用的解热镇痛类药物,同样可作为兽药使用。人体在过量摄取对乙酰氨基酚时有可能导致药物性肝损伤或急性肝衰竭[15],因此在现行国家标准中,要求对动物源性食品中的对乙酰氨基酚残留量进行检测[16]。
笔者将应用于血药分析领域的限进介质色谱法引入食品分析领域,简化了样品处理过程,在保证测量精确度的前提下,提高了分析效率。
1 实验部分
1.1 主要仪器与试剂
液相色谱仪:半微柱高效液相色谱仪Nanospace系统,配备二元高压泵、紫外检测器,日本大阪曹达株式会社。
电子天平:ML204/02 型,感量为0.01 mg,瑞士梅特勒-托利多公司。
离心机:GL-16C型,上海安亭科学仪器厂。
1.3.2 不良反应评价 根据1998年WHO抗癌药物不良反应表现及分级标准评估各组患者化疗过程中的不良反应,分为0~Ⅳ度,无毒性:0度;轻度毒性:Ⅰ~Ⅱ度;严重毒性:Ⅲ~Ⅳ度。
超纯水机:Milli-Q UltrapUre Ⅰon-ExTM型,美国MⅠLLⅠPORE公司。
震荡混悬器:MT-31型,日本YAMATO公司。固相萃取柱:PEP 30 mg/1 mL,美国艾杰尔-飞诺美公司。
乙腈、乙酸、乙酸铵:色谱纯,北京百灵威科技有限公司。
乙酸乙酯:色谱纯,国药集团化学试剂有限公司。
对乙酰氨基酚对照品:批号为C12328510,上海麦克林生化科技股份有限公司。
动物源性食品样品:猪肉,羊肉,牛肉:市售。
1.2 液相色谱条件
限进介质色谱柱:CAPCELL PAK MF Ph-1 柱[150 mm×2.0 mm,5 μm,三耀精细化工品销售(北京)有限公司];柱温:30 ℃;流动相:0.1%乙酸-0.05 mol/L乙酸铵溶液-乙腈(体积比为95∶5),等度洗脱,流量为0.3 mL/min;进样体积:20 μL;检测波长:250 nm。
1.3 实验方法
1.3.1 溶液制备
乙酸铵溶液:50 mmol/L,精密称取3.85 g 乙酸铵,精密量取1 mL乙酸溶液,用水定容至1 000 mL,经0.22 μm水膜过滤。
乙酰氨基酚标准储备溶液:1 mg/mL,精密称取对乙酰氨基酚对照品10 mg,置于10 mL容量瓶中,用甲醇溶解并稀释至标线。
系列乙酰氨基酚标准工作溶液:精密量取乙酰氨基酚标准储备溶液,用流动相稀释,配制成质量浓度分别为10、25、50、100、200、400、800、1 600 μg/L的系列标准工作溶液。
1.3.2 样品处理
将动物源性食品样品用研磨机充分粉碎,搅拌均匀,称取5 g 于具塞离心管中,加入20 mL 乙酸乙酯,超声10 min后涡旋2 min,然后以12 000 r/min离心10 min。取上清液至另一干净的离心管中,残渣加入10 mL 乙酸乙酯重复提取一次,再次离心后取上清液,合并上清液,于45 ℃水浴氮气吹干,用5 mL流动相复溶后,以12 000 r/min离心10 min,取上清液过0.22 μm滤膜,作为样品溶液。
1.3.3 样品测定
试样液经限进介质色谱柱高效液相色谱仪分析,测得峰面积,采用外标法通过上述标准曲线计算其浓度。
2 结果与讨论
2.1 色谱柱的选择
首先尝试使用GB 29683—2013[16]使用的常规反相C18色谱柱。结果表明,因其不具有在线排除大分子蛋白及油脂的功能,故在不进行SPE 净化时,容易出现蛋白堵塞色谱柱的问题。随后进行了不同键合相类型的限进介质色谱柱比较,键合相类型包括苯基键合、C8键合和磺酸基阳离子键合。由于目标物对乙酰氨基酚的化学结构可带正电荷,理论上通过反相机理或阳离子交换机理均可实现保留分离,但在实际试验后发现,C8键合型限进介质色谱柱对基质分离效果较差,样品中杂质对目标物存在干扰(图1);磺酸基阳离子键合型限进介质色谱柱对目标物的保留时间较短,样品中杂质对目标物存在干扰(图2);苯基键合型限进介质色谱柱对目标物的分离相对较好,且目标物保留时间理想(图3)。综合考虑分离和保留效果,最终选择苯基键合型限进介质色谱柱。
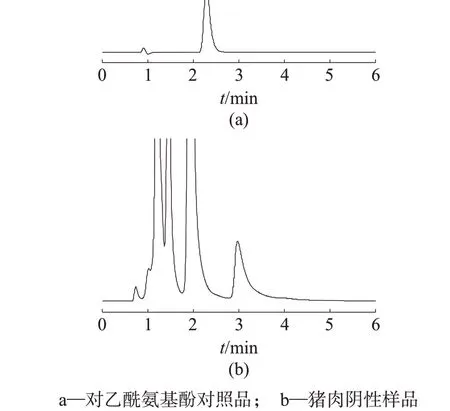
图1 使用C8键合型限进介质色谱柱的色谱图
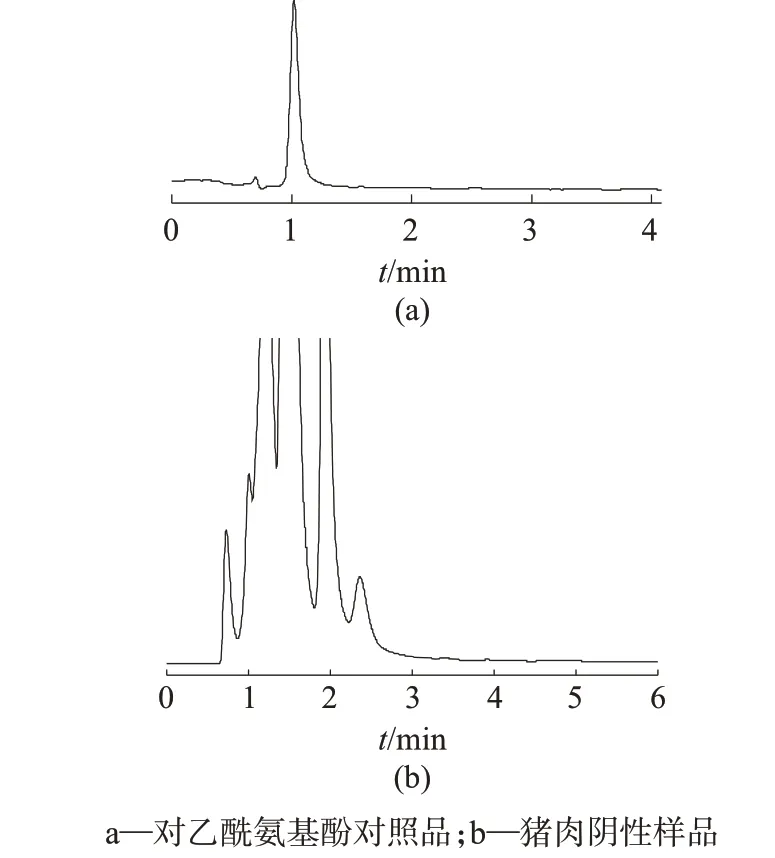
图2 使用磺酸基键合型限进介质柱的色谱图

图3 使用苯基键合型限进介质柱的色谱图
2.2 流动相的确定
GB 29683—2013使用C18色谱柱分析动物源性食品中的对乙酰氨基酚,流动相为0.05 mol/L 乙酸铵溶液-乙腈(体积比为8∶2)。换用限进介质色谱柱后,研究发现,增大水相比例有利于提高对乙酰氨基酚的分离度,并获得更大的响应值,当两相体积比为95∶5时效果最佳。此外,向流动相中添加质量分数为0.1%的乙酸后,在目标物响应值未变的基础上,获得了更稳定的保留时间。
选择10 μg/kg的加标样品,连续测定3天,每天重复分析3 次,计算色谱峰面积均值及保留时间的相对标准偏差。不同流动相种类及比例对目标物分析结果的影响如图4所示。
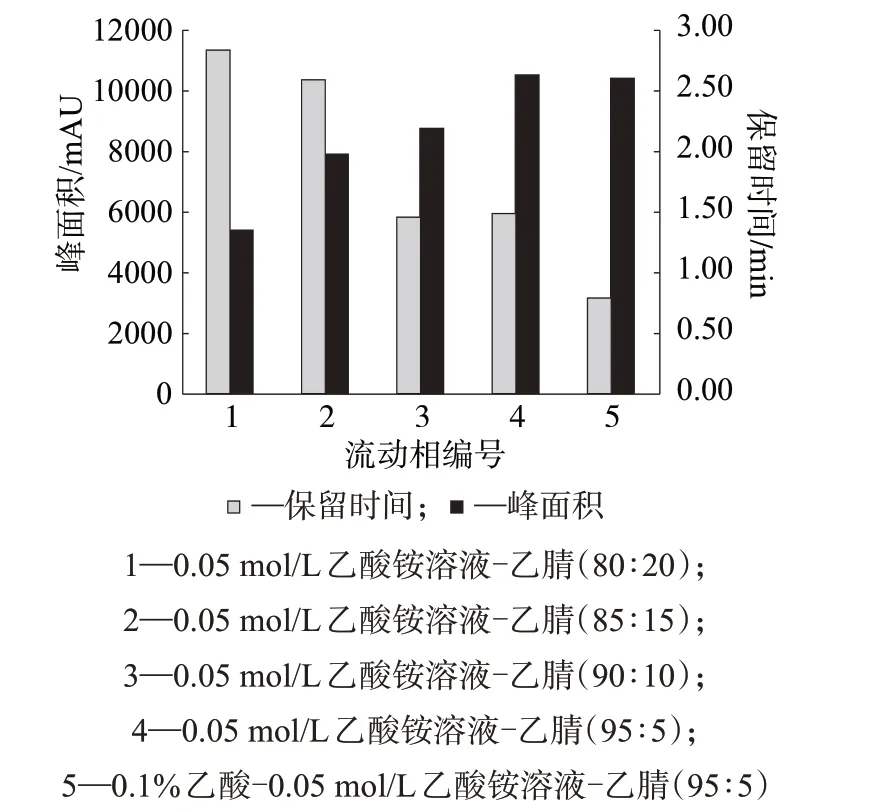
图4 使用不同流动相时乙酰氨基酚的色谱响应值
2.3 样品处理方法
GB 29683—2013中样品处理方法包括提取、除脂、净化3 个步骤,操作步骤繁多复杂,对乙酰氨基酚的回收率不高(回收率下限要求仅为70%)。提取过程是用于将动物源性食品中的对乙酰氨基酚提取释放,该过程不能省略,可考虑省略除脂、净化步骤。
笔者建立的方法省略了除脂、净化两个步骤,分别采用国家标准方法和该方法对添加质量分数为10 μg/kg 的猪肉样品进行处理后测定,6 次平行试验测定结果表明,国家标准方法对乙酰氨基酚的平均回收率及其相对标准偏差分别为78.2%、8.9%,该方法对乙酰氨基酚的平均回收率及其相对标准偏差则分别为99.3%、5.4%,明显优于国标方法。该样品处理方法不仅提高了乙酰氨基酚回收率,还节省了试剂耗材,大幅降低了样品处理成本,减少了分析时间。
2.4 色谱柱适用性考察
使用限进介质色谱柱对仅进行了液液提取的样品分析,考察色谱柱的寿命,结果表明,分析200份样品后色谱柱理论塔板数未见明显变化,表明该色谱柱可长期稳定使用。
2.5 线性范围、检出限和定量限
精密吸取系列乙酰氨基酚标准工作溶液各20 μL,注入液相色谱仪测定。以溶液质量浓度为横坐标,以相应的色谱峰面积为纵坐标,拟合标准曲线线性方程。结果显示,乙酰氨基酚质量浓度在10~1 000 μg/L范围内线性方程为y=189.808x-259.144,相关系数为0.999 9,表明线性关系良好。
以3倍信噪比对应的加标样品中乙酰氨基酚的质量分数作为方法检出限,以10倍信噪比对应的加标样品中乙酰氨基酚的质量浓度作为方法定量限。样品中对乙酰氨基酚的检出限和定量限分别为0.6、2 μg/kg,优于现行国家标准方法(检出限和定量限分别为3、10 μg/kg)。
2.6 样品加标回收试验
对猪肉阴性样品进行低、中、高3个浓度水平加标:将猪肉阴性样品分别使用研磨机充分粉碎并搅拌均匀后,分别称取样品5 g 于3 只具塞离心管中,分别加入0.1 μg/mL乙酰氨基酚标准溶液0.1、0.5、1 mL,再分别加入19.9、19.5、19 mL 乙酸乙酯,超声10 min后涡旋2 min,以12 000 r/min离心10 min,取上清液至另一干净的离心管中,残渣加入10 mL 乙酸乙酯重复提取一次,再次离心后取上清液,合并上清液,于45 ℃水浴氮气吹干,用5 mL 流动相复溶,以12 000 r/min离心10 min,取上清液过0.22 μm滤膜,进样测定。添加水平分别为2、10、20 μg/kg,每个添加水平进行6 次平行试验,加标回收试验结果见表1。
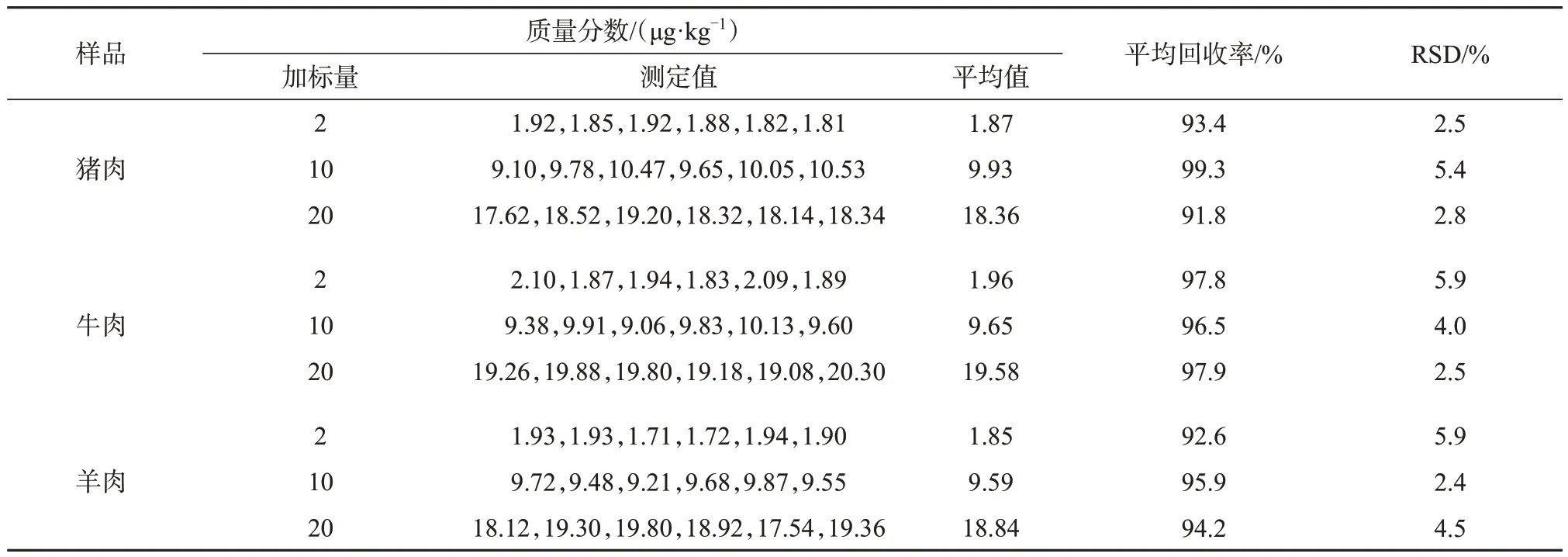
表1 样品加标回收试验结果
由表1 可知,3 种样品的平均加标回收率为85.3%~105.3%,相对标准偏差均小于5.9%,表明该方法具有较好的回收率和重现性。猪肉、牛肉、羊肉加标样品色谱图如图5所示。
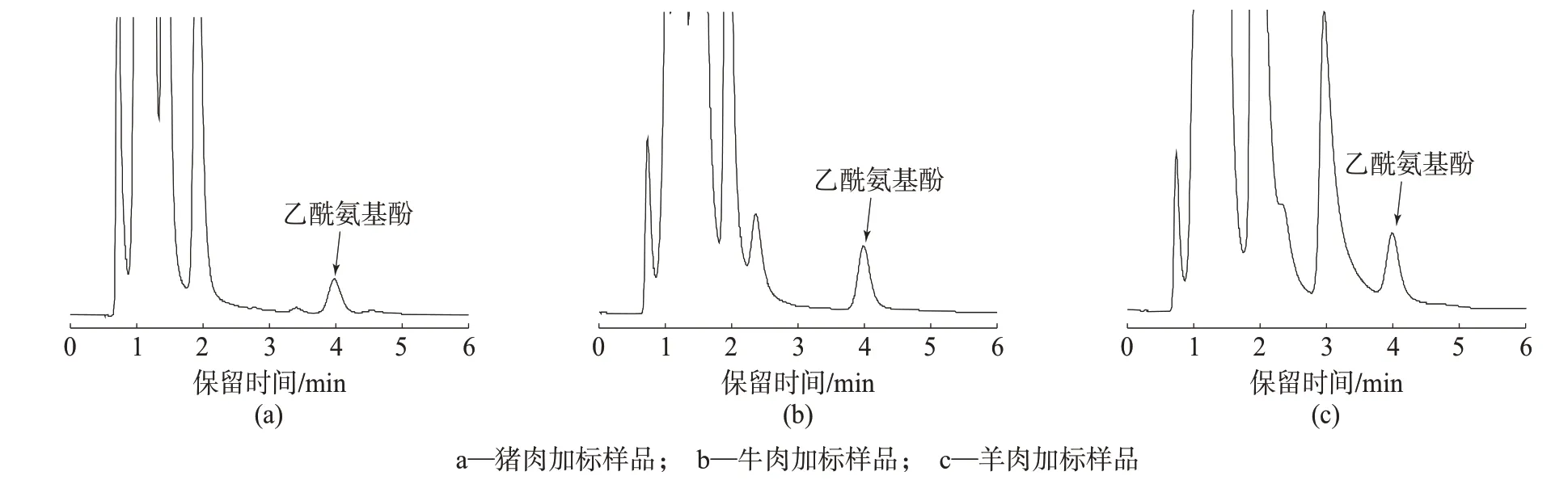
图5 加标样品色谱图
3 结语
将限进介质色谱法引入食品分析领域,可大幅简化样品前处理过程,大幅降低实验人员的时间成本和试剂耗材成本。该方法操作简单便捷,专属性强,灵敏度高。
