二十二碳六烯酸标准样品的研制
2023-12-07荣辉邓建朝陈胜军杨贤庆艾红闫帅江睿章丽萍
荣辉,邓建朝,陈胜军,杨贤庆,艾红,闫帅,江睿,章丽萍
(中国水产科学研究院南海水产研究所,农业农村部水产品加工重点实验室,国家水产品加工技术研发中心,广州 510300)
二十二碳六烯酸(DHA)是人体必需而又不能直接合成的多不饱和脂肪酸(PUFA),在促进婴幼儿视、脑神经发育,防治老年痴呆,改善人脑记忆及抗氧化,抗肿瘤等方面具有重要的生理调节功能[1-2]。DHA传统的商业来源主要为深海鱼油,由于渔业资源的日益紧张,加之鱼油提纯工艺复杂且产品大多具有难以根除的鱼腥味等,迫使人们渐渐把目光转向新的DHA 来源——微藻[3]。目前已发现多种微藻中富含PUFA[4-5],如裂壶藻、隐甲藻等。微藻是海洋食物链中PUFA 的最初生产者,有些藻细胞中DHA 含量较高,其相对含量可达细胞干重的5%~6%[6-7],而且所含的PUFA 种类比较单纯,进行单一成分的分离提纯相对容易一些,因而利用微藻生产多不饱和脂肪酸是一个非常有前景的商业领域[8]。
国外大多采用尿包法结合高效液相色谱法制备高纯度的DHA[9],该方法消耗较多的有机溶剂,提取工艺复杂,回收率低且成本过高。目前市场上高纯度的DHA标准样品主要来源于美国Sigma公司,不仅购货时间长,而且价格昂贵。因此急需开发一种简单有效,相对低廉的提取方法以满足人们对高纯度DHA 日益增长的需求。上世纪80 年代初,美国Ⅰto 教授[10]发明了高速逆流色谱(HSCCC),其流动相和固定相均为液体,很快被应用在医药学、生物化工、海洋生物学、食品科学、材料学等众多研究领域。近年来HSCCC 技术不断改进和提升,已被广泛应用在各种大分子物质的分离纯化研究中[11-13],如蛋白质、色素和脂肪酸等。Cao等[14]利用HSCCC分离葡萄籽油中的亚油酸,其纯度达到99%;孙磊等[15]利用HSCCC 法分离共轭亚油酸及其异构体,最终得到的共轭亚油酸及其异构体的纯度分别可达97.05%和97.73%。由此可知,利用HSCCC 技术分离纯化油脂中的不饱和脂肪酸是可行的。近年来,我国对不饱和脂肪酸类物质的应用开发成为热点[16],每年出口大量的不饱和脂肪酸产品,在国际市场上占有重要地位。但是,由于缺乏标样与标准,国产脂肪酸类物质多以化工原料出口,而科学研究、检测、药用标准品一直依赖国外进口,严重制约了与脂肪酸类物质相关的科学研究与医药产品的开发。
针对国内科研、生产对DHA 标准样品的需求,笔者依据GB/T 15000.3—2008 标准样品工作导则[17],利用HSCCC技术研制了DHA标准样品,可为含有DHA 相关产品的含量检测分析及质量控制提供技术保障。
1 实验部分
1.1 主要仪器与试剂
高速逆流色谱仪:TBE-300B型,上海同田生物技术有限公司。
气相色谱-质谱联用仪:GCMS-QP2010 Plus型,日本岛津公司。
高效液相色谱(HPLC)仪:LC-20AD 型,日本岛津公司。
紫外可见分光光度计:UⅤ 2550 型,日本岛津公司。
电感耦合等离子体质谱仪:Agilent 7900 型,美国安捷伦科技有限公司。
卡式水分仪:915Ti-touch型,瑞士万通公司。
红外光谱仪:FT-ⅠR NⅠCOLET 6700 型,美国热电公司。
离子淌度-Q-TOF 高分辨液质联用仪:Synapt G2-Si型,美国沃特世公司。
核磁共振波谱仪:Bruker AⅤANCE Ш HD 600型,德国布鲁克公司。
高效离心机:Avanti J-26XP 型,美国贝克曼库尔特有限公司。
冷冻干燥机:Alpha1-4型,德国Christ有限公司。
裂壶藻粉:利用松花粉垂钓法分离纯化获得的裂壶藻经28 ℃、180 r/min发酵培养96 h后,在4 ℃、转 速8 000 r/min 的 条 件 下 离 心 收 集 藻 泥,再 经-45 ℃真空冷冻干燥24 h后获得[18]。
细菌中性蛋白酶样品:(1) 1 600 U/g,Protex 7 L;(2) 580 000 U/g,Protex 6 L,杰能科(中国)生物工程有限公司。
DHA原料样品:纯度为99%,美国Sigma公司。
乙腈、甲醇、正己烷:色谱纯,广州华屿欣实验器材有限公司。实验所用其它试剂为分析纯。实验用水为一级水。
1.2 实验方法
1.2.1 裂壶藻油的提取和皂化
称取20 g 裂壶藻粉,以料液比(质量∶体积)1∶7的比例加入浓度为0.1 mol/L、pH 4.8的柠檬酸-柠檬酸钠缓冲液,匀浆均质处理5次后;再将藻液利用高通量组织研磨仪在55 Hz的条件下研磨150 s;调pH至9.5,在60 ℃恒温水槽中,匀速搅拌下碱提2 h;利用柠檬酸钠调反应体系pH至6.5,加入藻粉质量7%的中性蛋白酶,45 ℃恒温水浴条件下酶解3 h,再利用NaOH 调pH 至9.4,加入藻粉质量10%的碱性蛋白酶,68 ℃恒温水浴条件下酶解6 h;将酶解液在转速4 800 r/min条件下离心10 min后,静置分层,取上层游离油,放入真空干燥箱中在80 ℃条件下干燥2 h,冷却后称量。
称取500 mg裂壶藻油,加入0.5 mol/L的氢氧化钾-甲醇溶液25 mL,充分摇匀后在70 ℃恒温水浴条件下反应1.5 h,用20%硫酸水溶液调pH 至3;再用等量体积的乙醚萃取3次,静置分层,合并收集乙醚相,再通过旋转蒸发去除乙醚后,用HSCCC 两相溶剂体系正庚烷-乙酸乙酯-甲醇-水(15∶1∶15∶1,体积比,下同)的上下相各10 mL 溶解,得到裂壶藻脂肪酸粗样品,待利用HSCCC进行下一步的分离纯化。
1.2.2 HSCCC两相溶剂体系的选择
利用HPLC 测定DHA 原料样品在不同两相溶剂体系中的分配系数(K值),根据测得的K值选择最适的两相溶剂体系。先取适量的两相溶剂体系溶解DHA 标准品,再分别取上相和下相以HPLC 法检测,DHA在上相和下相中的色谱峰面积比值即为K值。HSCCC 两步分离法的两相溶剂体系分别为正庚烷-乙酸乙酯-甲醇-水(15∶1∶15∶1)和正庚烷-甲醇-水(5∶6∶1,体积比,下同)。
1.2.3 HSCCC法分离纯化DHA
先以10 mL/min的流量将两相容积体系的上相泵入仪器螺线管中,待充满后,将逆流色谱仪设为正转方式,转速为900 r/min,分离温度为13 ℃,再以3 mL/min的流量泵入两相容积体系的下相,以下相为流动相,待充分平衡后,取少量DHA 原料样品溶于流动相后进样分析,于210 nm 波长下进行紫外检测,以确定DHA的色谱保留时间。将皂化处理后获得的裂壶藻脂肪酸粗样,在同样的高速逆流色谱条件下进行两步法分离,对照DHA色谱保留时间收集相对应的组分并用HPLC 法进行纯度检测,最终将收集获得的DHA 高纯度样品通过旋转蒸发去除有机溶剂后,再经真空冷冻干燥,即为高纯度DHA 样品,该样品充氮气后密封保藏在-20 ℃冰箱。
1.2.4 纯度分析
(1) HPLC 法。收集分离纯化后的DHA 样品配制成质量浓度为0.25 mg/mL 的甲醇溶液,采用HPLC面积归一法测定其纯度。HPLC检测条件:进样体积为20 μL,色谱柱为C18(4.6 mm×250 mm,5µm,美国安捷伦科技有限公司)分析色谱柱,柱温为35 ℃,紫外检测器,检测波长为210 nm,流动相A为含0.05%(体积比)甲酸的超纯水,流动相B 为乙腈,恒定流量为1 mL/min。
(2)气相色谱-质谱(GC-MS)法。气相色谱条件:进样口温度为230 ℃,进样体积为1 μL;色谱柱为DB-5MS 色谱柱(0.25 mm×30 m,0.25 μm,美国安捷伦科技有限公司);采用程序升温,先于110 ℃保留4 min,再以10 ℃/min的速度升温至160 ℃,保留1 min,最后以5 ℃/min 的速度升温至240 ℃,保留15 min,载气为氦气,流量为1.52 mL/min,进样分流比为1∶30。
质谱条件:质量扫描范围为m/z40~550,电子能量为70 eⅤ,离子源温度为200 ℃,切除溶剂保留时间为3 min。
1.2.5 杂质分析
(1)水分分析。采用卡式水分仪利用卡尔费休库仑滴定直接进样法[19]测定DHA 样品中的水分含量,精确称取高纯度DHA 样品10.0 mg,测定3 次,取平均值。
(2)无机元素分析。将DHA 样品配成质量浓度为0.1 mg/mL的甲醇溶液。利用电感耦合等离子体质谱(ⅠCP-MS)法[20]测定Li、Be、B、Na、Mg、Al、Si 等无机元素的含量,测定3次,取平均值。
1.2.6 结构确认
采用紫外光谱(UⅤ)、红外光谱(ⅠR)、质谱(MS)和核磁共振波谱(NMR)等技术对制备获得的DHA 样品进行结构确认和表征,具体分析条件分别为:(1)紫外光谱采用紫外可见分光光度计对样品进行扫描,扫描范围为200~400 nm;(2)红外光谱采用液体样品制片法,扫描波数为450~4 000 cm-1;(3)质谱为电喷雾离子源,电子能量为70 eⅤ,离子源温度为200 ℃,扫描范围为100~1 000;(4)核磁共振以氘代甲醇为溶剂。
1.2.7 分装与储存
将制备的DHA 样品分装在2 mL 棕色样品瓶中,分装是在超净工作台中进行的,以每瓶10 mg分装,共分装100 瓶。将分装好的样品置于一个可以密封的塑料盒中,放置在-20 ℃冰箱中保存。
1.2.8 均匀性分析
按照标准样品工作导则要求,随机抽取10瓶分装后的DHA样品,每瓶样品平行测定3次。先用少量甲醇溶解DHA样品,最终配制成质量浓度为0.25 mg/mL的DHA甲醇溶液,采用HPLC面积归一法测定DHA样品纯度,利用方差分析法对纯度分析结果进行统计检验。
1.2.9 稳定性分析
短期稳定性:考察制备的DHA样品在4 ℃储存条件下放置9 天内的稳定性。每3 天取3 个平行样品采用HPLC 面积归一法测定其纯度,每个样品均测定3 次,取平均值,样品纯度的最终检测结果为3个平行样品的平均值。
长期稳定性:考察制备的DHA样品在-20 ℃储存条件下放置2 年内的稳定性。每6 个月取3 个平行样品采用HPLC 面积归一法测定其纯度,检测方法同上。
1.2.10 定值
根据GB/T 15000.3—2008 标准及JJF 1006—1994《一级标准物质技术规范》[21]相关规定和要求,最终选取8 家实验室对DHA 样品的纯度进行联合定值。从分装好的100 瓶DHA 样品中随机抽取24瓶,向每家实验室随机寄送3瓶,各实验室收到样品后先将其配制成质量浓度为0.25 mg/mL 的甲醇溶液,再采用HPLC面积归一法对其进行纯度定值,每个样品平行测定2 次,取平均值并计算标准偏差和扩展不确定度。
2 结果与分析
2.1 分离纯化
对所提裂壶藻油脂进行皂化处理后,在流动相流量为3 mL/min、高速逆流转速为910 r/min、分离温度为13 ℃的条件下利用HSCCC技术进行两步分离纯化,第一步的两相溶剂体系为:正庚烷-乙酸乙酯-甲醇-水(15∶1∶15∶1),测定K值为0.84;第二步的两相溶剂体系为:正庚烷-甲醇-水(5∶6∶1),测定K值为0.88。最终分离结果见图1,可获得高纯的DHA样品。
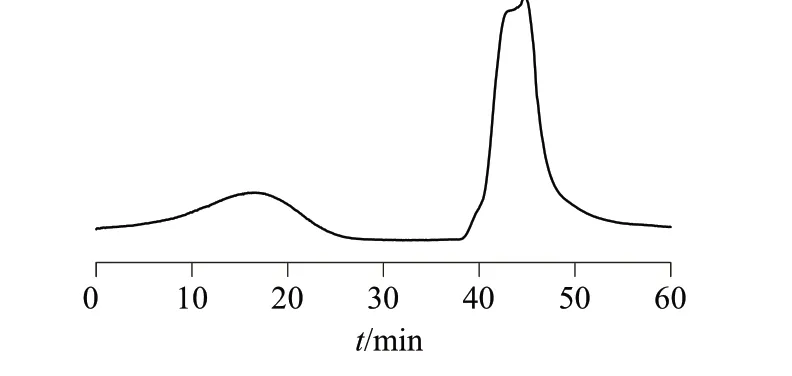
图1 HSCCC分离DHA色谱图
2.2 纯度分析
2.2.1 高效液相色谱法
利用HSCCC技术分离获得的DHA样品按20%流动相A 和80%流动相B 以恒定流量1 mL/min 进行HPLC等度洗脱40 min,色谱图见图2。扣除溶剂色谱峰以后,未发现明显杂质峰,以面积归一法定量,最终分离获得的DHA样品经高效液相色谱法测得的HPLC纯度为99.12%。
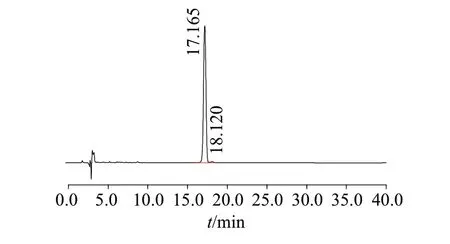
图2 DHA样品的HPLC纯度分析色谱图
2.2.2 GC-MS法
将制备的DHA 样品先经甲酯化后再进行GCMS 法纯度分析,以正己烷经同样甲酯化后作为空白对照,所测得的总离子流色谱图见图3,采用面积归一法定量,测定其纯度为99.18%。
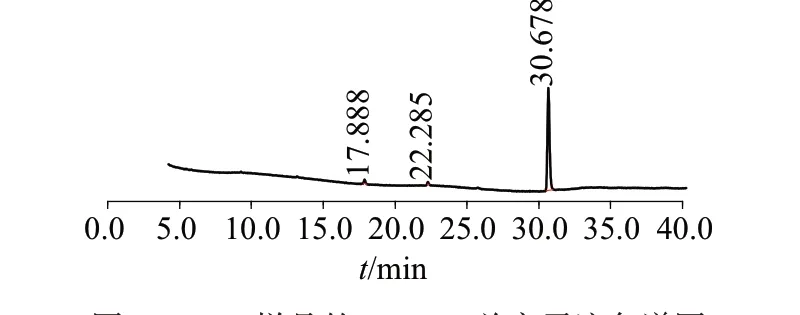
图3 DHA样品的GC-MS总离子流色谱图
2.2.3 水分分析
采用卡尔费休库伦电位滴定法直接测定DHA标准样品中的水分含量,测量3 次水分质量分数的平均值为(0.241±0.012)%。
2.2.4 无机元素分析
将DHA 样品配成质量浓度为0.1 mg/mL 的甲醇溶液。利用ⅠCP-MS 法测定Li、Be、B、Na、Mg、Al、Si 等无机元素的含量,3 次测定的平均结果为:Li、Be、B、Na、Mg、Al、Si、S、Cl、K,Ca、Sc、Ti、Ⅴ、Cr、Mn、Fe、Co、Ni、Cu、Zn、Ga、Ge、As、Se、Br、Kr、Rb、Sr、Y、Zr、Nb、Mo、Ru、Rh、Pd、Ag、Cd、Ⅰn、Sn、Sb、Te、Ⅰ、Cs、Ba、La、Ce、Pr、Nd、Sm、Eu、Gd、Tb、Dy、Ho、Er、Tm、Yb、Lu、Hf、Ta、W、Re、Os、Ⅰr、Pt、Au、Hg、Tl、Pb、Bi、Th 和U的含量和为(0.000 032 4±0.000 000 5)%。
2.3 结构分析
2.3.1 紫外光谱分析
通过紫外可见分光光度计扫描,可知制备的DHA 标准样品的最大吸收波长在200 nm 处左右,由于处于紫外吸收波长临界端,可能影响结果的稳定性,因此实际检测选取吸收值较最大处稍低的210 nm,DHA样品的紫外光谱图见图4。
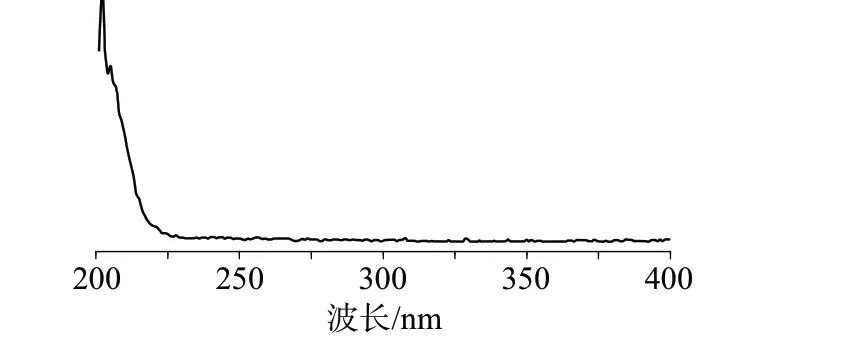
图4 DHA样品的紫外光谱图
2.3.2 红外光谱分析
先将制备的DHA样品进行液膜压片处理,再利用红外光谱仪对其进行扫描,扫描波长范围为450~4 000 nm。结果显示,711 cm-1为顺式烯烃上C—H的变形振动,1 711 cm-1为羧基的C=O 伸缩振动,2 965 cm-1为烷烃的C—H伸缩振动,3 014 cm-1为烯烃的C=C 双键伸缩振动,这些均为DHA 的特征峰。样品的红外光谱吸收波长数据均存在这些特征基团,并与DHA文献数据相符[22]。
2.3.3 高分辨质谱分析
采用电喷雾电离源(ESⅠ)负离子模式和Resolution分析模式,质量扫描范围为m/z50~1 000,获得丰度最大的离子峰为327.232 7[M-H]-,推断DHA 的相对分子质量为328.232 7。
2.3.4 核磁共振分析
以氘代甲醇为溶剂,DHA 样品1H-NMR 和13CNMR数据见表1。
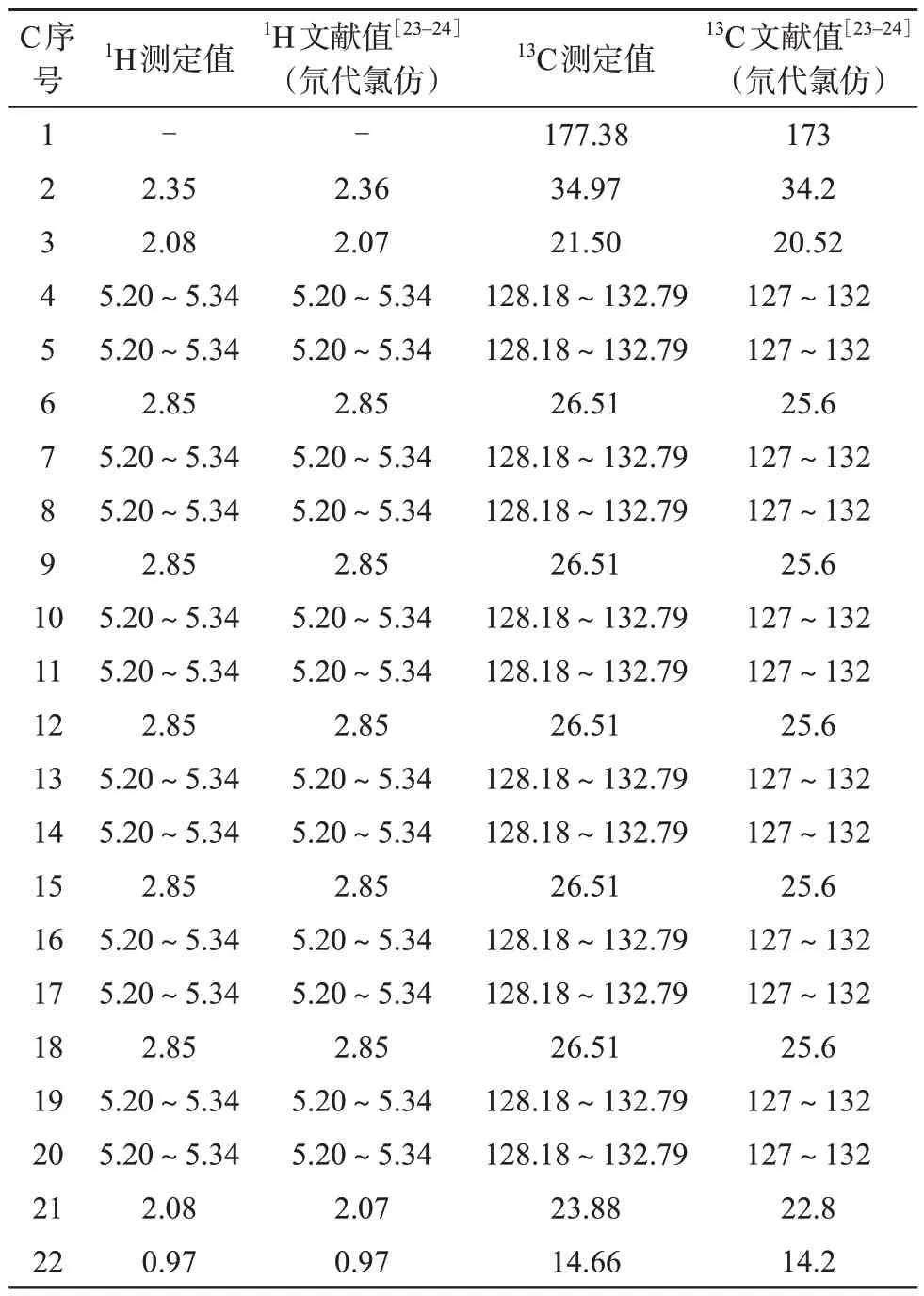
表1 DHA样品的核磁共振波谱数据(氘代试剂:氘代甲醇)
根据1H-NMR 谱显示由化学位移可以确认有5个CH2分别位于5 个烯键中间,另有1 个—CH2—CH2—分子片段与—C=O相连。根据13C-NMR谱,确定有7 个不饱和度,由12 个烯碳CH (6 个不饱和度)和1 个羰基碳C (1 个不饱和度)组成。分子中除去C=O,—CH2—CH3和6个—CH=CH—的分子结构片段,还有7个亚甲基。综上所述,样品的核磁共振谱图所给出的结构信息与化合物DHA 的化学结构式相符[23-24],故鉴定为DHA。
2.4 均匀性分析
DHA 标准样品的HPLC 纯度分析结果经F检验,以确定样品的均匀性数据是否符合正态分布,结果见表2和表3。
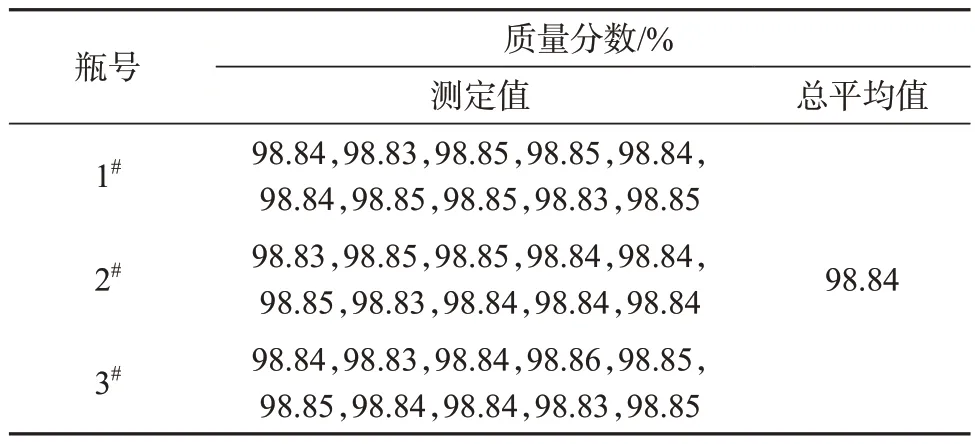
表2 DHA标准样品均匀性分析结果
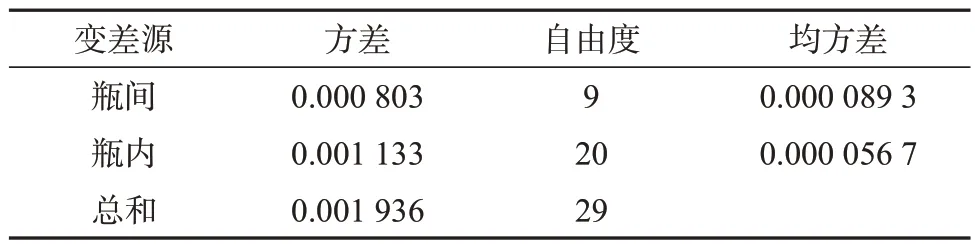
表3 DHA标准样品纯度方差分析结果
样品瓶间方差:sbb2=1.086 7×10-5,样品瓶间标准偏差:sbb=0.003 3,重复性标准偏差:sr=0.007 5,均匀性检验的不确定度ubb=sbb=0.003 3。查F界值表可知F0.05(9,20)=2.94,而根据DHA 样品数据计算所得F=1.57<F0.05(9,20),则说明该DHA标准样品是均匀的。
2.5 稳定性分析
2.5.1 短期稳定性
考察DHA 样品短期稳定性的数据见表4。由表4 可知,所测DHA 样品纯度平均值结果,统计分析差异不显著,测得的纯度平均值在所测时间内均未随时间的改变而明显升高或降低,表明该样品在4 ℃储存条件下9天时间内是稳定的。
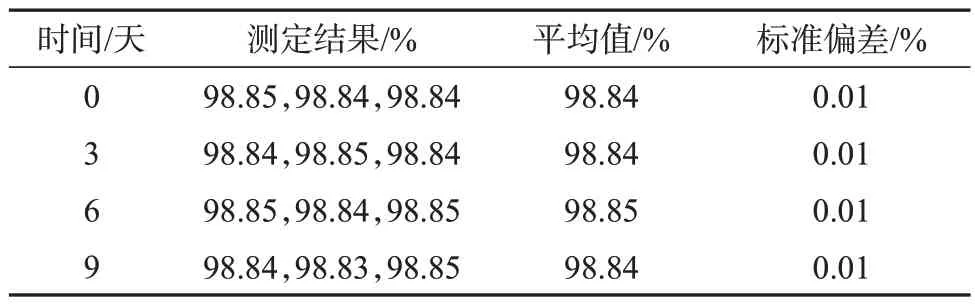
表4 短期稳定性试验结果
2.5.2 长期稳定性
DHA 样品长期稳定性考察数据见表5。从表5可看出,所测得的样品纯度平均值在所测时间内均未随时间的变化而显著升高或降低,纯度测定值保持在(98.84±0.02)%的范围内,表明该样品在-20 ℃储存条件下2年时间内是稳定的。
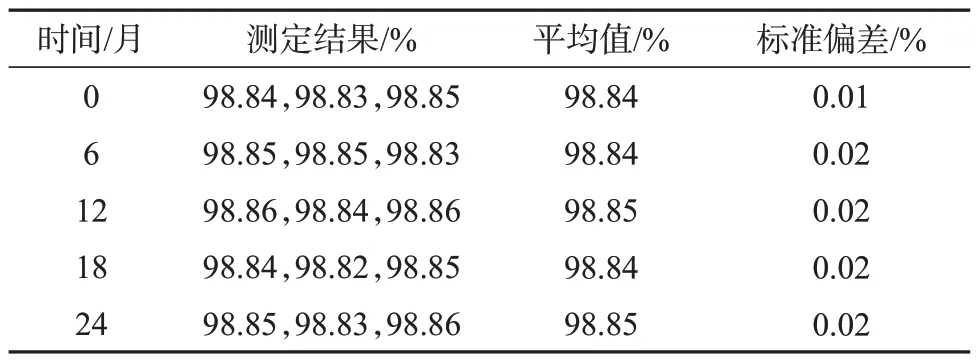
表5 长期稳定性试验结果
采用t检验法统计分析DHA 样品的纯度数据,利用直线作经验模型,根据直线斜率变化是否显著,预测样品的稳定性。计算结果为:截距b0=98.84,直线斜率b1=0.000 3,斜率标准偏差s(b1)=0.000 274。当自由度为n-2=3和P=0.95 (95%置信水平)时经查表可知,t(0.95,3)=3.18,而|b1|=0.000 3<t(0.95,3)×s(b1)=0.000 87,说明直线斜率变化不显著,因此样品在-20 ℃储存条件下2年时间内稳定性良好。
2.6 定值
由8家实验室采用HPLC面积归一法对DHA样品纯度进行联合定值,对定值的统计分析结果见表6。
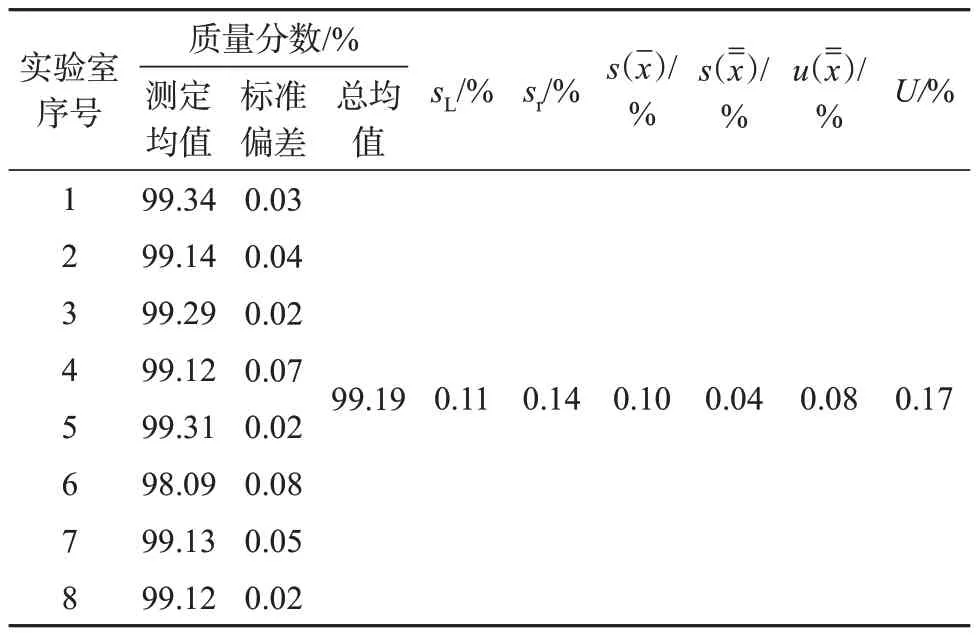
表6 DHA样品HPLC纯度定值的统计分析结果
根据标准样品工作导则规定及研制报告要求,由8家实验室联合定值试验引入的标准不确定度为总平均值的不确定度:u(--x)=0.08;由样品均匀性引入的标准不确定度为:ubb=sbb=0.003 3,由样品稳定性引入的标准不确定度为:ults=2×s(b1)×t=0.013 2,则DHA样品定值引入的扩展不确定度为:
DHA样品的HPLC纯度测定值为99.19%,扣除水分和无机元素含量,最终DHA样品纯度的定值结果为98.95%,扩展不确定度为0.17% (k=2,P=0.95)。
3 结语
采用HSCCC技术制备获得高纯度的DHA样品并根据GB/T 15000.3—2008《标准样品工作导则》规定及要求研制DHA标准样品并进行纯度定值,定值结果为98.95%,置信度95%的扩展不确定度为0.17% (k=2,P=0.95)。研制的DHA标准样品可用于含有DHA相关产品的含量检测分析和质量控制等。
