脑出血后小胶质细胞极化及其相关炎症信号通路对继发性脑损伤的影响
2023-12-04孔德敏邹伟
孔德敏,邹伟
脑出血是一种极具破坏性的卒中亚型,致残率和死亡率都很高,全球每年约有280万人因脑出血死亡,约占卒中相关死亡的50%。尽管脑出血后脑损伤机制的研究已取得显著进展,但目前在临床上脑出血诱发的继发性脑损伤(secondary brain injury,SBI)仍缺乏有效的治疗方法[1-3]。研究表明,脑出血后发生SBI的潜在机制包括炎症、神经元死亡(包括凋亡和坏死)、铁诱导损伤、线粒体功能障碍、氧化应激和活性氧的产生等[4],各机制之间存在相互联系,但尚未得到充分阐明。小胶质细胞活化在脑出血中的双重作用体现在抑炎型小胶质细胞发挥吞噬作用促进神经功能恢复,促炎型小胶质细胞释放炎症因子加重炎症反应,加速SBI病情进展。小胶质细胞极化可通过不同信号通路来干预脑出血后SBI病情的发展。因此,本文主要阐释小胶质细胞与脑出血诱发SBI的关联以及对调控小胶质细胞极化的相关通路机制进行研究,为脑出血后SBI的治疗靶点提供理论依据并重新思考脑出血的治疗方向。
1 小胶质细胞与脑出血继发性脑损伤
小胶质细胞在成人脑细胞中占5%~10%,并形成中枢神经系统(central ner vous system,CNS)中最大的免疫细胞群,可维持CNS环境的免疫平衡[5-6]。小胶质细胞具有高度多样化,促炎型小胶质细胞分泌促炎因子导致CNS恢复受到抑制,而抑炎型小胶质细胞释放抗炎介质减缓组织炎症损伤,促进组织的再生与修复,两者对脑出血后神经炎症反应的调控及病情进展有决定性影响[7]。脑出血患者通常出现进行性恶化,提示SBI在脑出血的病理机制中起着关键作用[8]。SBI主要以原发性脑损伤、血肿血液成分释放、神经胶质细胞激活和血源性免疫细胞渗入脑部为特征。当大脑内稳态被破坏时,小胶质细胞作为大脑常驻的免疫细胞和脑损伤的第一反应者会被激活并发挥调节作用。脑出血后小胶质细胞数量的增加可能是由于损伤诱导的小胶质细胞增殖或小胶质细胞迁移到损伤部位以及血源性巨噬细胞的脑浸润。SBI常导致血脑屏障破坏、脑水肿、血肿扩大、血肿周围细胞死亡、严重的神经功能恶化以及神经行为功能预后不良,这均与小胶质细胞激活息息相关[9]。研究表明,脑出血后,小胶质细胞的激活在3 d达到峰值,7 d后显著下降,尽管白细胞浸润在2~3 d后逐渐消退,但这些激活的小胶质细胞所诱导的SBI仍然存在,促炎型小胶质细胞过度释放破坏性的促炎介质和神经毒性物质,导致血脑屏障完整性破坏、血肿周围水肿和神经功能障碍,抑炎型小胶质细胞可促进脑出血后血肿的内源性清除[10-12]。因此,促炎与抑炎型小胶质细胞间的转化可能是对脑出血诱发SBI的一种潜在治疗方式。
2 调控促炎型小胶质细胞极化的信号通路
2.1 TLR4/NF-κB信号通路 Toll样受体(Toll-like receptor,TLRs)是参与先天免疫的经典分子家族,与CNS疾病的炎症反应密切相关,Toll样受体4(Toll-like receptor 4,TLR4)作为TLR家族中参与先天免疫的重要成员,主要表达于小胶质细胞或星形胶质细胞中[13-14],而其下游核转录因子κB(nuclear factor kappa-B,NF-κB)在响应损伤后,从细胞质转入细胞核,释放趋化因子、细胞因子和其他细胞毒性化学物质调节炎症反应[15]。脑出血后TLR4/NF-κB-p65通路会被激活,其介导的免疫调节作用和神经炎症反应有助于星形胶质细胞活化,而活化的星形胶质细胞可以使小胶质细胞向促炎表型极化,并分泌大量的炎症因子,进而通过神经炎症反应加重SBI[16-17]。因此,它已成为脑出血治疗的常见抗炎靶点。研究表明,当TLR4沉默时,在脂多糖(lipopolysaccharide,LPS)刺激的BV-2小胶质细胞中,乔松素仍然能够下调NF-κB-p65的磷酸化并增加抑炎型小胶质细胞的数量。这一结果表明,乔松素主要通过抑制TLR4激活和小胶质细胞促炎型极化来减轻脑出血后SBI的病情进展[18]。另外,孤核受体4A1(nuclear receptor subfamily 4 group A member 1,NR4A1)在小胶质细胞中可负向调节NF-κB的转录活性,抑制炎症基因的表达。美拉诺坦(nle4-D-Phe7-α-MSH,NDP-MSH)通过促进小胶质细胞抑炎表型极化发挥抗炎作用,在大鼠小胶质细胞中NDP-MSH治疗有助于上调NR4A1,下调MMP-9和炎症相关分子,从而减弱脑出血后的神经炎症反应和血脑屏障的破坏[19]。
2.2 Tim-3/Gal-9信号通路 T细胞免疫球蛋白和黏蛋白结构域蛋白-3(T cell immunoglobulin and mucin domaincontaining protein 3,Tim-3)作为一种新的免疫调节分子,广泛表达于先天免疫细胞中,并在免疫调节和耐受中发挥复杂作用[20]。在CNS疾病中Tim-3的表达水平升高使小胶质细胞激活,并在与中性粒细胞浸润相关的炎症过程中起着至关重要的作用[21]。半乳糖凝集素-9(galectin 9,Gal-9)在免疫耐受和炎症反应中至关重要,Gal-9作为Tim-3的配体可被Tim-3上调,参与与炎症反应相关的CNS疾病[22]。在脑出血急性期,Tim-3与Gal-9结合可促进小胶质细胞向促炎表型极化,释放促炎因子,加剧SBI,故阻断Tim-3/Gal-9通路已被证明可减缓脑出血后的SBI[23]。研究表明,Tim-3在人类和小鼠的先天免疫系统细胞上均具有组成性表达,可以与TLRs协同作用[24]。脑出血后,Tim-3的高水平表达诱发了两种炎症通路的激活,其中TLR4信号通路的激活与小胶质细胞向促炎表型转化关系密切;Tim-3/Gal-9信号通路的激活亦可促进炎症因子的释放,加重脑出血后的SBI。因此,全面了解Tim-3、Gal-9和TLRs之间的关系可以更好地解释脑出血后SBI的病理机制。
2.3 JNK/p38MAPK信号通路 丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPKs)是细胞内的一类丝氨酸/苏氨酸蛋白激酶,其亚型包括p38MAPK、c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)和细胞外信号调节激酶(extracellular regulated protein kinases 1/2,ERK1/2),是调节由某些刺激诱发炎症反应的关键信号分子。脑出血后,MAPK信号通路被激活的作用之一是支持小胶质细胞在血肿区存活,从而确保小胶质细胞对神经元的细胞毒活性,故调节MAPK和小胶质细胞活性是预防脑出血诱发SBI进展的重要策略[25]。研究表明,脑出血后JNK/p38MAPK信号通路被激活,使促炎介质(TNF-α、IL-6和IL-1β)被释放和神经元细胞死亡[26]。JNK/p38MAPK信号通路调节NOD样受体热蛋白结构域相关蛋白(NOD-like receptor thermal protein domain associated protein,NLRP)炎性小体和IL-1β在外周免疫细胞、缺血初级皮质神经元和缺血条件下的脑组织中表达,NLRP3炎性小体主要表达于小胶质细胞,使小胶质细胞向促炎表型转化并产生促炎细胞因子,诱导炎性事件的发生[27]。实验表明,在仔猪脑出血模型的白质束中可发现小胶质细胞促炎表型和促炎介质的表达增加,并导致MAPK信号通路的急性激活,使用米诺环素治疗后出现转化生长因子-β(transforming growth factor-β,TGF-β)的表达上调和p38MAPK的激活降低,并抑制促炎型小胶质细胞的表达,从而减轻脑出血后神经炎症诱发的白质损伤[28]。
2.4 N O D1/R I P 2 信号通路 核苷酸结合寡聚结构域1(nucleotide-binding oligomerization domain 1,NOD1)是NOD样受体家族的一员,作为胞浆内先天免疫受体,在感知病原体或组织损伤时,可驱动先天免疫反应,并触发炎症级联反应[29-30]。此外,NOD1及其下游分子受体相互作用蛋白2(receptor interacting protein 2,RIP2)在调节NF-κB和MAPK信号激活中发挥关键作用且与神经炎症相关[31-32]。在脑出血中,NOD1和RIP2表达显著增加,并促进了小胶质细胞激活和炎症反应,抑制NOD1或RIP2的表达可防止脑出血诱导的SBI。这种保护作用的机制可能是防止小胶质细胞从静止状态向过度激活状态转变,抑制促炎型小胶质细胞释放相关炎症因子(包括诱导型一氧化氮合酶、IL-1β和TNF-α)。然而,NOD1/RIP2与炎症因子IL-1β和TNF-α之间存在窜扰,它们之间存在正反馈且相互激活,这在一定程度上导致脑出血过程中有持续的炎症反应并加剧SBI,揭示了NOD1/RIP2信号通路是对抗脑出血相关神经炎症的重要潜在治疗靶点[33]。
2.5 JAK2/STAT3信号通路 Janus激酶2(janus kinase 2,JAK2)/信号转导和转录激活因子3(signal transducer and activator of transcription 3,STAT3)通路是近年新发现的一条细胞内信号转导途径,与抑制炎性反应、神经退行性病变及脑出血等CNS疾病的病理过程密切相关[34]。JAK2/STAT3通路是小胶质细胞极化的重要信号转导通路,可通过STAT3和NF-κB通路促进炎症反应,使小胶质细胞向促炎型分化[35]。研究发现,细胞因子信号转导抑制因子3(suppressor of cytokine signaling 3,SOCS3)是STAT3信号通路的主要负调控因子,增强SOCS3的表达,可抑制炎症因子的释放,并通过负向调控JAK/STAT和NF-κB-p65信号通路使小胶质细胞向抑炎型转化,进而减轻脑出血早期的炎症反应[36]。
2.6 PKC δ/CARD9信号通路 PKC δ作为新型蛋白激酶C(protein kinase C,PKC)亚型家族的一员,是炎症反应的关键调节因子,且在脑出血后PKC δ的表达增加[37],而胱天蛋白酶募集域蛋白9(caspase recruitment domain protein 9,CARD9)通过NF-κB信号通路启动炎症细胞因子级联并招募中性粒细胞浸润[38]。通过动物实验发现,PKC δ/CARD9是介导髓系细胞触发受体-1(triggering receptor expressed on myeloid cells-1,TREM-1)影响脑出血后小胶质细胞极化的关键胞内蛋白通路,可促进小鼠脑出血后早期炎症反应的发生[39],故抑制PKC δ/CARD9信号通路介导的TREM-1可使小胶质细胞从促炎型向抑炎型极化,减少神经炎症,从而改善脑出血后神经行为的结局。
3 调控抑炎型小胶质细胞极化的信号通路
3.1 PI3K/AKT/mTOR信号通路 哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)是一种丝氨酸/苏氨酸激酶,亦是磷脂酰肌醇3激酶(phosphatidylinositol 3 hydroxykinase,PI3K)相关激酶家族的一员。PI3K/蛋白激酶B(protein kinase B,AKT)信号通路的激活已被证明可以减轻脑损伤、抑制神经炎症和神经元凋亡,而mTOR作为PI3K/AKT通路下游的重要靶点,其通路被激活可以诱导损伤神经元合成蛋白质,从而促进广泛的轴突再生[40-41]。脑出血后微小核糖核酸29a(microRNA 29a,miR29a)表达升高,并通过激活PI3K/AKT/mTOR通路促进轴突生长和神经恢复[42]。研究指出,磷酸酶及张力蛋白同源物基因(phosphatase and tensin homologous gene,PTEN)作为PI3K的拮抗剂,可以抑制PI3K/AKT/mTOR信号通路的启动;相反,抑制PTEN可激活PI3K/AKT/mTOR通路,减轻体内出血性脑损伤和体外细胞毒性诱导的神经元损伤,还可激活同侧海马中的PI3K/AKT通路,有效缓解脑出血诱导的继发性海马损伤,从而改善迟发性认知缺陷[43]。miR-23b通过负向调控多磷酸肌醇激酶(inositol polyphosphate multikinase,IPMK)的表达水平,进一步激活AKT/mTOR自噬通路,抑制脑出血后诱发的神经炎症反应并发挥神经功能保护作用,使其成为治疗脑出血的潜在靶点[44]。众学者对mTOR介导的自噬通路机制阐述不一,未来仍需对该通路上下游各种信号分子的功能做进一步的研究。
3.2 ERK/Nrf2/HO-1信号通路 ERK/核因子E2 相关因子2(nuclear fact orerythroid 2-related factor 2,Nrf2)/血红素氧合酶1(heme oxygenase-1,HO-1)信号通路是体内重要的抗炎抗氧化信号通路,与脑出血后神经损伤修复密切相关。研究显示,Nrf2通路激活可减轻小胶质细胞所造成的炎症反应,ERK1/2已被证明是C-C趋化因子受体4(C-C chemokine receptor 4,CCR4)激活后的下游蛋白,活化的CCR4可促进ERK1/2磷酸化,Nrf2被磷酸化的ERK1/2激活后进入细胞核,促进受Nrf2调控的下游靶蛋白HO-1转录,在脑损伤时HO-1在小胶质细胞中的表达增加,而Nrf2通过增加抑炎型小胶质细胞CD163和CD36的表达来促进脑出血后血肿的消退,故ERK/Nrf2/HO-1信号通路可作为治疗脑出血诱发SBI炎症反应的潜在药物靶点[45-46]。
3.3 BDNF/TrkB信号通路 脑源性神经营养因子(brain-derived neurotrophic factor,BDNF)是一种重要的神经营养因子,能够促进神经元的生长、发育及成熟。酪氨酸激酶受体B(tyrosine Kinase receptor B,TrkB)属于神经营养受体家族中的一员,主要在哺乳动物大脑中表达。BDNF作为体内含量最多的神经营养因子,与TrkB结合形成BDNF/TrkB通路,该通路已被广泛认为是脑损伤后修复的关键通路[47]。研究表明,脑出血后抑炎型小胶质细胞中BDNF的表达增加,进而增加TrkB磷酸化,触发大量信号蛋白的激活,使血肿周围的微环境改变,促进神经元修复,发挥脑出血后神经保护作用[48]。
3.4 miR-575/PTEN信号通路 研究表明,在脑出血红细胞损伤后,L-赖氨酸增加了受损皮质神经元中miR-575的表达,而miR-575表达增加可减少受损皮质神经元的损伤[49]。PTEN作为miR-575的下游靶基因,与炎症反应密切相关,抑制PTEN的表达可促进小胶质细胞向抑炎表型极化。L-赖氨酸亦可促进抑炎型小胶质细胞极化并减少炎症反应,在脑出血损伤中发挥神经保护作用,而这是因为脑出血损伤后miR-575(上调)/PTEN(下调)信号通路的介导。
3.5 PPARγ信号通路 过氧化物酶体增殖物激活受体γ(peroxisome proliferatorsactivated receptor γ,PPARγ)是一种重要的转录因子,在开启促炎或抑炎标记基因中起着至关重要的作用,并介导抑炎型小胶质细胞极化的启动[50]。PPARγ激活可介导下游蛋白CD36的转录,作为抑炎型小胶质细胞的生物标志物,有助于吞噬能力的体现。脑出血后通过PPARγ信号通路的激活可使抑炎型小胶质细胞极化,加速血肿吸收并改善神经功能预后[51]。
4 脑出血诱发继发性脑损伤的药物靶点治疗
目前,对于一些治疗脑出血有前景药物的主要潜在机制是抑制促炎型小胶质细胞的激活和增强抑炎型小胶质细胞的抗炎作用。乔松素、瑞莎托维是TLR4信号通路的抑制剂;米诺环素对MAPK信号通路有抑制作用,但最新研究发现米诺环素通过BDNF/TrkB途径促进抑炎型小胶质细胞的极化,并促进BDNF和神经元细胞的生成;依达拉奉右莰醇是一种新型的神经保护剂,具有抗氧化和抗炎的协调效应,通过抑制TLR4/NF-κB信号通路可以更好地减少炎症因子的表达,控制炎症反应;芬戈莫德在LPS活化的小胶质细胞中通过负向调控p38MAPK和NF-κB信号通路可明显减少促炎型小胶质细胞的激活,降低大脑炎性反应;雷帕霉素作为mTOR通路的激动剂,可增加抑炎型小胶质细胞的表达,减少神经炎症反应带来的脑损伤[52-56]。图1表明了调控促炎型、抑炎型小胶质细胞极化的信号通路和相关药物作用靶点之间的关系。
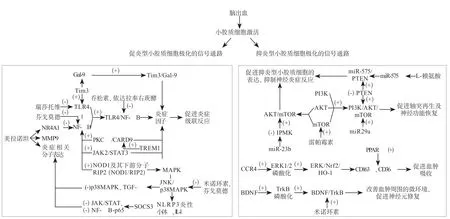
图1 小胶质细胞极化的信号通路和相关药物作用靶点之间的关系Figure 1 Relationship between the signal pathway of microglia polarization and related drug action targets
5 总结与展望
综上所述,在脑出血诱发SBI中小胶质细胞起着“双刃剑”的作用,其治疗的时间窗口需要进一步明确。早期干预促炎型小胶质细胞向抑炎型转变对于促进血肿清除、减轻早期脑出血后水肿形成、促进神经功能恢复可能具有一定的临床意义,但尚需进一步实验以明确实施干预的可行性及价值。调节小胶质细胞表型不仅可能加速血肿的吸收,还可能改善白质完整性、脑修复和功能恢复,这可能有助于改善脑出血后的长期神经行为结局。小胶质细胞激活所介导的促炎反应是神经炎症的关键步骤,通过一系列信号转导途径靶向小胶质细胞促炎表型极化过程,可为临床抗炎治疗提供可能性。另外,有“清道夫细胞”之称的小胶质细胞可吞噬外来物质和细胞碎片,在脑出血血液清除中起着关键作用,减少了血肿的有害机械效应,防止由血液成分(包括铁)诱导的细胞毒性。在整个病理进程中,参与的调控因子以及各种信号通路十分庞杂,本文所探讨的仅是这个过程的冰山一角。小胶质细胞极化在脑出血导致的SBI中的作用机制仍可能会发现新的治疗靶点,从而改善脑出血的预后。目前,脑出血的治疗方案主要集中在减少血肿扩大和预防并发症上。尽管进行了一些随机临床试验,但迄今为止,还没有药物或手术疗法可以显著改善脑出血后的人体功能结局。因此,促使小胶质细胞表型的转化并面向临床,以及更全面地掌握不同信号通路和其中关键靶点的相互作用,是下一步的研究方向。
利益冲突所有作者均声明不存在利益冲突。
