以精神行为异常为突出表现的遗传性脑小血管病1例报道
2023-12-04陈嫄周玉颖李攀
陈嫄,周玉颖,李攀
HtrA丝氨酸蛋白酶1(HtrA serine protease-1)又称高温必需因子A-1(hightemperature requirement protease A1,HTRA1),其基因纯合或复合杂合突变可导致伴皮质下梗死和白质脑病的常染色体隐性遗传性脑动脉病(cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy,CARASIL),是临床罕见的单基因隐性遗传性脑小血管病(cerebral small vessel disease,CSVD),临床主要表现为脱发、脊椎退行性病变、卒中、痴呆、情感障碍等[1]。近年来,国内外先后报道HTRA1基因杂合突变同样具有致病性,其突变的基因型与临床表型存在相关性[2]。位于17号染色体q21上的GRN基因编码颗粒蛋白前体(progranulin,PGRN),突变的GRN基因被认为是额颞叶痴呆常见的致病基因,但是近年来的研究表明其临床表型异质性很强。GRN基因突变会导致PGRN水平降低,增加痴呆风险[3]。目前对于中国人HTRA1及GRN基因突变的临床表型均认识不足。本文报道1例以精神行为异常为突出表现的遗传性CSVD,基因检测结果提示HTRA1基因杂合突变,同时伴GRN基因杂合突变,希望此病例能为今后深入研究提供线索。
1 病例介绍
患者男性,58岁,司机,初中学历,身高174 cm,体重73 kg,BMI 24.1 kg/m2,主因“记忆力下降1年,性格改变、行为异常6个月,反应迟缓、定向力下降2个月”于2018年1月2日就诊于天津市环湖医院神经内科认知障碍门诊。患者于1年前无明显诱因出现记忆力下降,以近记忆力下降为主,有时不能回忆刚刚发生的事,重复提问刚刚问过的事情;有时买东西不能算对账;家人未予重视。患者于6个月前出现性格改变及行为异常:脾气暴躁,总因一点小事发脾气;性格变得非常固执,总按自己的意愿行事,不顾及他人感受,不听从他人建议;与陌生人交谈时好像认识对方,过度交谈;白天困倦,晚上失眠;认为家人让他吃药是害他;反复冲马桶,拾捡垃圾。患者于入院前2个月,两次在家门口迷路,无法自己找回家;去熟悉的地方坐错公交车;反应迟缓,不愿与家人交流。遂就诊于认知障碍门诊。
既往高血压病史7年,血压最高达160/100 mmHg(1 mmHg=0.133 kPa),平素口服硝苯地平(30 mg,1次/日)降压治疗,平素血压维持在130/80 mmHg。否认糖尿病、冠心病病史。脑梗死病史1个月,无明显后遗症。否认肝炎、结核、疟疾等传染病病史;否认外伤史;否认输血史;否认食物、药物过敏史。吸烟史20年,10支/日;偶尔饮酒。父母均有记忆力下降的病史,已过世;哥哥60岁时曾出现迷路现象,已过世;大姐38岁时患脑梗死,已过世;二姐40岁时患脑梗死,已过世;弟弟50岁时患脑梗死。否认其他家族遗传病病史。
体格检查:体温36.2 ℃,脉搏56次/分,呼吸18次/分,血压156/81 mmHg。胸廓无畸形,双肺呼吸音清,未闻及干湿 音。心音有力,心律齐,各瓣膜听诊区未闻及病理性杂音。腹软,无压痛,肝脾肋下未触及,双下肢不肿,脊柱、四肢无畸形。神经科查体:神清,主动语言减少,时间、地点、人物定向力可,计算力减退,近记忆力下降,双侧瞳孔等大,左∶右=3 mm∶3 mm,光反应(+),眼动可,眼位居中,无复视及眼震,左侧鼻唇沟浅,伸舌居中,软腭上抬正常,咽反射(+),四肢肌力Ⅴ级,肌张力正常,腱反射(++),双侧巴宾斯基征(-),双侧感觉检查对称,双侧共济检查稳准。
辅助检查:血常规、凝血四项(血浆凝血酶原时间、活化部分凝血活酶时间、凝血酶时间、纤维蛋白原)、免疫全项、肝肾功能、血脂、血糖、血流变、超敏CRP、Hcy、甲状腺功能、血液三项(叶酸、维生素B12、铁蛋白)、风湿免疫全项(甲型肝炎、乙型肝炎、丙型肝炎、梅毒及艾滋病)、尿常规、便常规均未见明显异常。心电图示窦性心律,正常心电图。心脏彩色超声示二尖瓣、三尖瓣轻度反流。腹部、泌尿系彩色超声检查未见异常。神经心理学评估结果见表1。
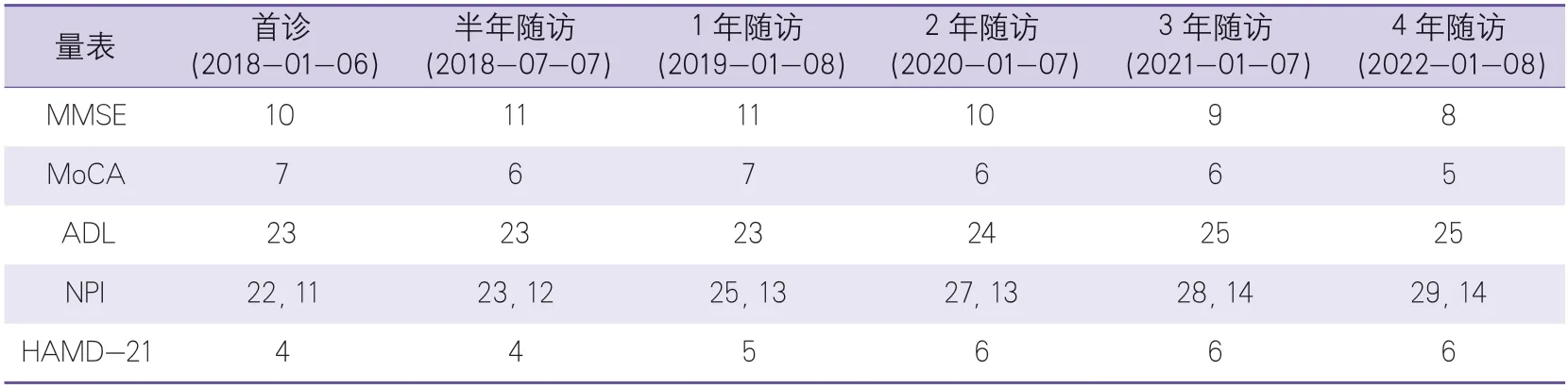
表1 患者神经心理量表评估结果Table 1 Evaluation results of neuropsychological scales of the patient单位:分
头颅MRI(2018-01-04)示右侧脑室枕角旁DWI稍高信号,考虑梗死灶;双侧小脑、脑桥、双侧丘脑区、双侧基底节区、双侧脑室旁、双侧半卵圆中心、双额及胼胝体腔隙灶及软化灶;双侧基底节区、双侧丘脑区、右侧颞枕交界、双侧脑室旁、左额及右顶点状梯度回波序列(gradient-recall echo,GRE)低信号,考虑含铁血黄素沉积、缺血性脑白质脱髓鞘改变,脑白质病变Fazekas分级3级(图1A~E)。头颅MRA(2018-01-04)示左侧大脑后动脉P2段局限性狭窄;右侧大脑后动脉P2段局限性纤细(图1F)。患者及其儿子和女儿行全外显子组基因测序,结果示患者及其女儿均携带HTRAl基因杂合突变(chr10:124221201,c.34delinsTCCT;chr10:124221646,c.472+6C>A);患者及其儿子和女儿均携带GRN基因杂合突变(chr17:42426891,c.236G>A),生物信息学软件SIFT和Polyphen2预测该变异分别为有害的和很可能有害的变异,目前该突变位点尚未被报道,基因报告建议判定为临床意义不明确(variants of uncertain significance,VUS)变异(图2)。脊椎MRI检查(2018-02-18)示颈椎和腰椎退行性病变。
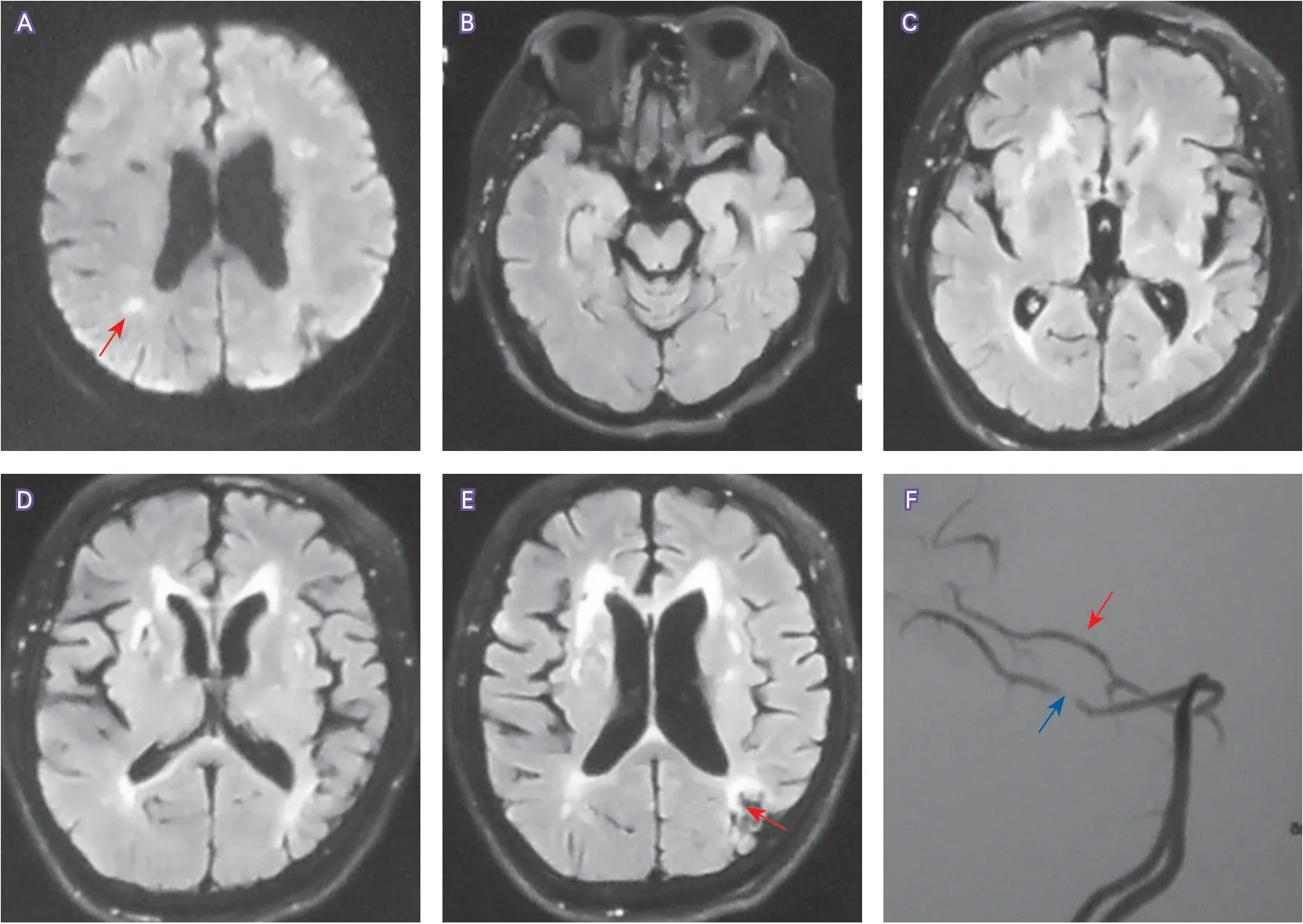
图1 患者头颅MRI和MRA结果(2018-01-04)Figure 1 Results of MRI and MRA for patient’s head(2018-01-04)

图2 患者及其子女基因检测结果Figure 2 Gene testing results for the patient and his children
治疗和随访:给予阿司匹林0.1g 1次/日,丁苯酞0.2克/次,3 次/日,美金刚5 m g 1次/日,1周后调整至10 mg 1次/日,奥拉西坦0.8克/次,3次/日,氨氯地平5 mg 1次/日,口服治疗。6个月及1年对患者随访发现,患者认知功能、日常生活能力较前无明显变化,患者对家里的事和家人漠不关心,情感淡漠。在妄想、激越、脱抑制、睡眠障碍方面较前无明显变化,而在易激惹、异常的运动行为方面程度较前增加。2年、3年、4年对患者随访发现,患者认知障碍程度缓慢进展,在妄想、激越、易激惹、脱抑制、异常行为、睡眠障碍等方面程度较前严重(表1),尚无新发急性脑梗死。
2 讨论
CARASIL是1965年日本学者首先报道的一种罕见的单基因隐性遗传性CSVD[4],青年期即可发病,临床上以脱发、反复卒中发作、进行性认知水平下降及腰痛为主要临床表现,同时颈椎、腰椎MRI可见椎体和椎间盘退行性改变[5]。遗传学研究表明CARASIL的致病基因为位于10q26染色体的HTRA1基因,该基因编码HTRA蛋白酶合成。HTRA1基因纯合或复合杂合突变,导致HTRA1蛋白酶活性丧失,不能抑制转化生长因子(transforming growth factor,TGF)-β家族信号转导,使纤维连接蛋白的蛋白结构域A(extra domain A,EDA)和多功能蛋白聚糖在小动脉内膜中积累增厚,导致管腔狭窄、动脉硬化和血管重塑,进而发生缺血性卒中,同时TGF-β调控头皮毛囊发育及骨骼形成,导致脊椎病和脱发[6]。
近年来,HTRA1基因杂合突变被发现同样与CSVD相关,HTRA1基因杂合突变在常染色体显性CSVD中的致病作用已被证实。目前其分子致病机制尚不完全明确,既往多项家族性CSVD基因研究分析发现,不同位点、部位、突变类型都可导致HTRA1蛋白酶活性受损,HTRA1蛋白酶活性丧失或降低是HTRA1基因杂合突变导致CSVD的先决条件[2,7-8]。其潜在机制可能是由于部分错义突变为占优势的负突变,突变的等位基因由于缺乏活性位点而失去活性,成为功能缺失突变体,干扰剩余野生型等位基因正常功能,使酶活性进一步下降;也可能是由于某些错义突变为半显性突变,或是由于无义介导的mRNA或不稳定蛋白的降解,突变等位基因的HTRA1蛋白表达量减少,从而导致单倍功能不全[9]。另外,诱导HTRA1基因由非活性晶体结构向活性结构变化的L3/LD环结构域发生突变,使HTRA1蛋白酶的活性降低,进而影响酶活性激活的信号传导,也可能是另一潜在的致病机制[10]。
既往关于C S V D人群的队列研究发现,不同国家和地区HTRA1基因杂合突变在家族性CSVD人群中发病率有所差异,日本人群发病率为6.52%[7],欧洲人群发病率为4.9 8%[7-8](其中意大利人群发病率为3.52%[11]),中国台湾省汉族人群的发病率为5.61%[12]。HTRA1基因杂合突变不仅在家族性CSVD人群中被发现,在散发性CSVD患者中也有报道。Verdura等[7]发现在日本67例散发性CSVD患者中有3例存在HTRA1基因杂合突变。在中国台湾省无家族史的115例CSVD患者中发现1例HTRA1基因杂合突变和NOTCH3突变[12]。HTRA1基因杂合突变相关CSVD的临床表现、影像学特征与CARASIL相似,但两者也存在差异。Uemura等[2]对既往文献报道的HTRA1基因相关的CSVD总结发现,HTRA1基因杂合突变相关CSVD患者的确诊年龄晚[(59.8±10.5)岁],临床症状较轻,神经系统症状(认知障碍、卒中、步态障碍等)出现较晚[(54.0±11.4)岁]。与CARASIL相比,HTRA1基因杂合突变相关CSVD缺血性卒中的发生率更高(63%),而脱发(13.2%)、步态障碍(67.4%)、脊椎硬化和退行性变(颈椎、腰椎为主)(60%)发生率相对更低。
本例患者为中老年男性,无脱发,有家族缺血性卒中及认知障碍史,临床以认知障碍起病,进行性加重,表现为记忆力下降,随着疾病进展出现性格改变、行为异常,神经系统体格检查提示高级智能减退,左侧中枢性面瘫。头颅MRI主要表现为侧脑室枕角旁亚急性梗死灶、多发腔隙性梗死灶及软化灶、多发微出血;脑白质病变Fazekas分级3级。脊椎MRI提示颈椎和腰椎退行性病变。由于患者的父母、哥哥、大姐、二姐均已过世,弟弟不愿进行基因检测,因此只完善了患者及其子女的基因检测,结果提示患者及其女儿HTRAl基因在1号外显子(c.34delinsTCCT)和1号内含子(c.472+6C>A)发生杂合变异。该变异位点目前尚未见文献报道。患者发病年龄晚,无脱发,有缺血性卒中及认知障碍表现及相关家族史,颈椎和腰椎发生退行性病变,头颅MRI符合典型CSVD影像学改变,支持既往关于HTRA1基因杂合突变相关CSVD研究的临床表型。然而HTRA1基因杂合突变相关CSVD病例报道中关于患者精神症状的描述甚少,在已报道的中国台湾省9例汉族病例中,仅有4例患者存在强迫行为、抑郁和妄想等精神症状[12],而在日本、法国、意大利的相关病例报道中尚未有关于患者精神症状的详细描述。本例患者以认知障碍起病,随后以精神行为异常为突出表现,存在妄想、激越、脱抑制、睡眠障碍,且随着时间推移,精神症状持续加重。
GRN基因编码PGRN,可促进神经元和小胶质细胞的发育和存活。目前已鉴定出GRN基因中大约存在114个致病突变,一般多认为以单倍体功能不足为致病机制[13]。GRN基因功能丧失突变可引起不同临床表型,以额颞叶痴呆最为常见,包括阿尔茨海默病、帕金森症、皮质基底节综合征、进行性核上性麻痹、肌萎缩侧索硬化等[14-15]。与西方国家相比,GRN基因突变在中国较为罕见,在国内某些单一中心仅占1.2%~2.6%。既往报道的中国GRN基因突变携带者中,包括5例额颞叶痴呆、2例皮质基底节综合征、1例原发性进行性失语、1例非典型帕金森病和1例肌萎缩侧索硬化[13]。本例患者及其儿子和女儿均携带GRN基因杂合突变,位于3号外显子236G位碱基发生G>A的错义突变,尽管患者头颅MRI未发现GRN基因突变患者常见的明显的额颞叶不对称性萎缩,但患者精神行为异常突出,妄想、激越、脱抑制、睡眠障碍等精神症状持续进展加重,与行为变异型额颞叶痴呆临床表现相似,推测GRN的错义突变可能与该患者的发病机制相关,GRN基因与HTRA1基因协同作用导致该患者突出的精神行为异常。
3 结论
本文报道了1例以精神行为异常为突出表现的遗传性CSVD患者,扩展了与HTRA1基因相关的临床表型。HTRA1基因相关遗传性CSVD的基因与临床表现呈高度相关,对于家族性和散发性CSVD患者均应重视基因筛查。本例患者检出的GRN错义突变,由于未能完善家系验证尚不能确定其致病性,对于该家系成员长期随访以及完善该位点的功能验证将有利于明确GRN基因突变的致病性。本病例在多种基因相互作用影响临床表型方面有一些提示,也体现了在临床实践中基因与临床表型的关系的复杂性。
利益冲突所有作者均声明不存在利益冲突。
