基于高通量测序技术观察哮喘患儿上呼吸道菌群多样性的变化
2023-11-24秦大妮马铁梁沈惠平雷勇魏萍蒋益戴标
秦大妮, 马铁梁, 沈惠平, 雷勇, 魏萍, 蒋益, 戴标
(江苏大学附属宜兴医院儿科, 江苏 宜兴 214200)
哮喘是儿童常见的慢性呼吸系统疾病,发病机制目前尚未明确,与外周环境、宿主遗传和免疫机制密切相关[1-2]。呼吸道黏膜表面的正常菌群,随着儿童生长发育伴随宿主免疫功能和外界环境的影响不断完善,对维持呼吸道功能稳定起着重要作用[3]。研究发现,支气管黏膜上定植的菌群组成失调,即菌群间平衡被打破,可影响呼吸道正常功能,进而诱导哮喘的发作[4]。呼吸道菌群微生态的稳定有助于拮抗气道炎症反应,减轻哮喘发作频率[5]。目前基于16S核糖体RNA(16S rRNA)基因序列多态性的独立菌群分析,已在临床研究中得到广泛应用[6-7]。本研究拟通过高通量基因组测序技术,分析哮喘儿童呼吸道病原菌的分布特点及菌群类型,为进一步探索哮喘的个性化治疗提供依据。
1 资料与方法
1.1 研究对象
按照2016年儿童支气管哮喘诊断与防治指南的诊断标准[8],前瞻性纳入2020年10月至2021年10月宜兴市人民医院儿科收治的20例支气管哮喘患儿。纳入标准:选择学龄前哮喘儿童急性发作期入哮喘组。排除标准:① 慢性活动性肺部疾病患儿;② 纳入研究前4周有哮喘急性发作需要全身使用糖皮质激素;③ 肺功能测定前6 h吸入糖皮质激素和(或)短效支气管舒张剂;④ 呼出气一氧化氮(fractional exhaled nitric oxide,FeNO)测定前4 h进行剧烈运动;⑤ FeNO测定前进食菠菜、动物内脏等含高氮食物,测定2 h内有饮用可乐等刺激性饮料;⑥ 未同时完成FeNO及呼吸道菌群检测。其中,8例患儿未能完成FeNO测定,4例患儿合并重症感染未完成菌群检测,最终8例纳入本研究。将此8例支气管哮喘患儿设为哮喘组,30例健康体检学龄前儿童设为对照组,最终完成抽血及咽拭子采集共同完成者共18例。所有患儿来院就诊时采集咽拭子标本2份,同时抽取外周血2 mL备用。收集患者基本信息,检测白细胞(white blood cell,WBC)、中性粒细胞(neutrophil,NE)、淋巴细胞(lymphocyte,LY)、嗜酸性细胞(eosinophil,EO)、免疫球蛋白E(immunoglobulin E,IgE)及FeNO等指标。本研究已通过江苏大学附属宜兴医院医学伦理委员会审核批准(伦审2022科166),参加研究的儿童均得到监护人的知情同意,并签订知情同意书。
1.2 方法
1.2.1 上呼吸道样本的收集与DNA提取 在清洁环境中采集18例健康体检儿童和8例支气管哮喘患儿的上呼吸道标本,每例采集不少于2份,样本立即放入-80 ℃冰箱中冻存备用。按照DNA提取试剂盒(天根生化科技有限公司)说明书提取DNA,将提取的总DNA进行完整性检测,并用NanoDrop检测DNA的浓度,检测合格的DNA放入-20 ℃冰箱冻存。
1.2.216SrRNA基因V3V4区扩增与测序结果分析 应用Illumina Novaseq 6000平台对扩增产物进行高通量测序,由上海欧易生物医学科技有限公司完成。首先使用cutadapt软件,对raw data序列进行拆分,剪切引物序列。然后使用DADA2,将上一步合格的双端raw data,按照QIIME 2(2020.11)默认参数进行质控分析,得到扩增子序列变异(amplicon sequence variants,ASVs)丰度表格,使用Silva(version138)数据库比对,物种注释使用q2-feature-classifier软件进行分析,并对菌群稀释性曲线、测序质量、Alpha多样性、Beta多样性、菌群组成及菌群差异进行统计分析。
1.3 统计学处理
2 结果
2.1 两组临床基线特征及临床指标比较
与对照组比较,哮喘组在性别及年龄方面比较,差异均无统计学意义(P>0.05),见表1。与对照组比较,哮喘患儿WBC、NE及LY计数均无显著差异,但EO计数、IgE及FeNO明显高于对照组(P<0.05或P<0.01),见表2。
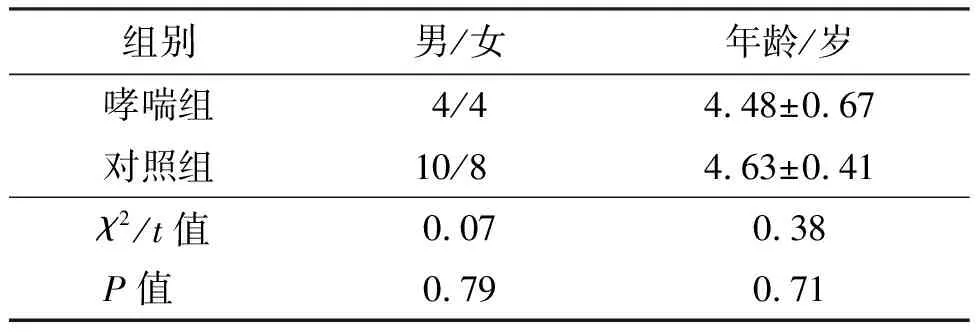
表1 对照组和哮喘组临床基线特征
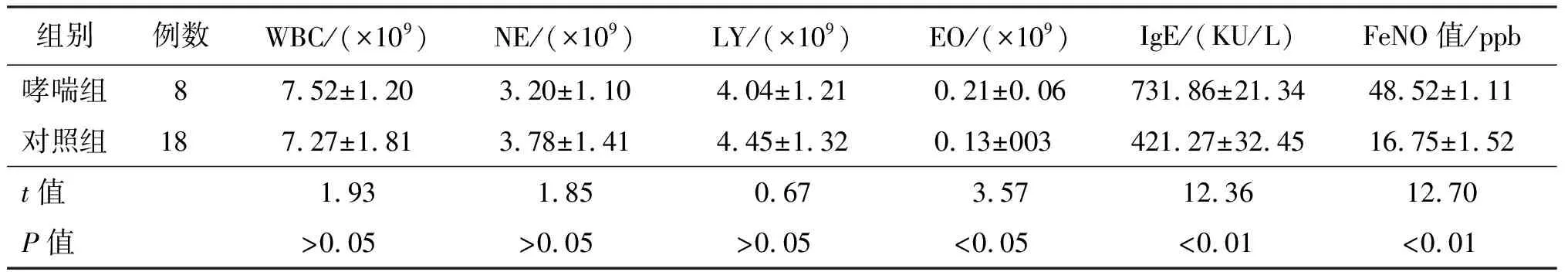
表2 对照组和哮喘组临床指标对比
2.2 菌群稀释性曲线
PD_whole_tree指数和Chao1指数均显示,当样本量逐渐增加到一定量后曲线趋向平坦(图1),说明测序数据量合理,更多的数据量只会产生少量新的ASVs。因此,稀释性曲线可得出样品有足够的数据量,并达到样品的测序深度。
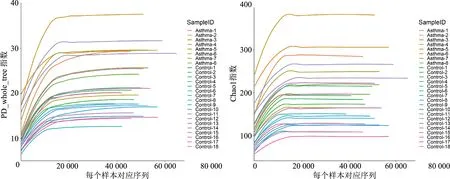
图1 ASVs多样性稀释曲线
2.3 测序质量统计
将收集到的18例对照组及8例哮喘组共26个样本进行16S rDNA测序,利用QIIME 2软件对原始序列进行过滤,哮喘组和对照组最终分别得到78 072和81 842条高质量数据,有效率分别为72.09%和84.00%,平均读长在405~424 bp之间。在100%的相似性水平下对有效序列进行ASVs聚类,结果显示,哮喘组和对照组共有ASVs 375个,特有ASVs分别为889和1 037个,哮喘组菌群ASVs相较于对照组发生显著变化。
2.4 Alpha多样性分析
哮喘组中反映群落丰富度的指标ACE指数和Chao1指数均明显高于对照组(P均<0.05),提示哮喘组中物种丰富度较大;哮喘组中反映群落多样性的指标Shannon指数的值明显高于对照组(P<0.05),Simpson指数的值亦有所增高,但差异无统计学意义(P>0.05),见图2。由此可见,哮喘组群落的多样性和物种丰富度明显高于对照组。

图2 Alpha多样性分析
2.5 Beta多样性分析
PCoA分析结果显示(图3),哮喘组与对照组的组内样本距离接近,组间样本点基本分开,表明哮喘组与对照组上呼吸道菌群组成发生明显变化(P<0.01)。
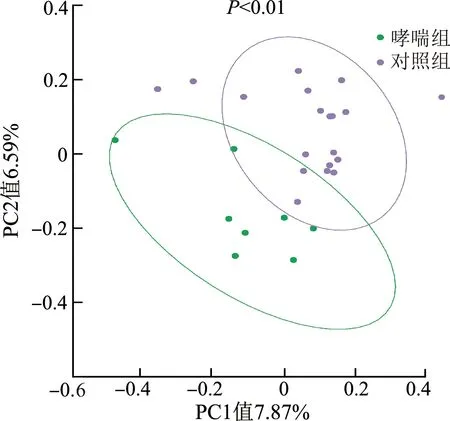
图3 PCoA分析结果
2.6 群落结构和丰度分析
如图4所示,在门水平,对相对丰度排在前15的主要菌门进行分析,结果表明哮喘组和对照组中的优势菌门主要为变形菌门、厚壁菌门、拟杆菌门、放线菌门、梭杆菌门、弯曲杆菌,总占比分别为98.33%和98.63%;与对照组相比,哮喘组放线菌门、梭杆菌门、弯曲杆菌门的相对丰度明显增加(P<0.05),厚壁菌门的相对丰度明显下降(P<0.01),而拟杆菌门的相对丰度无明显变化。
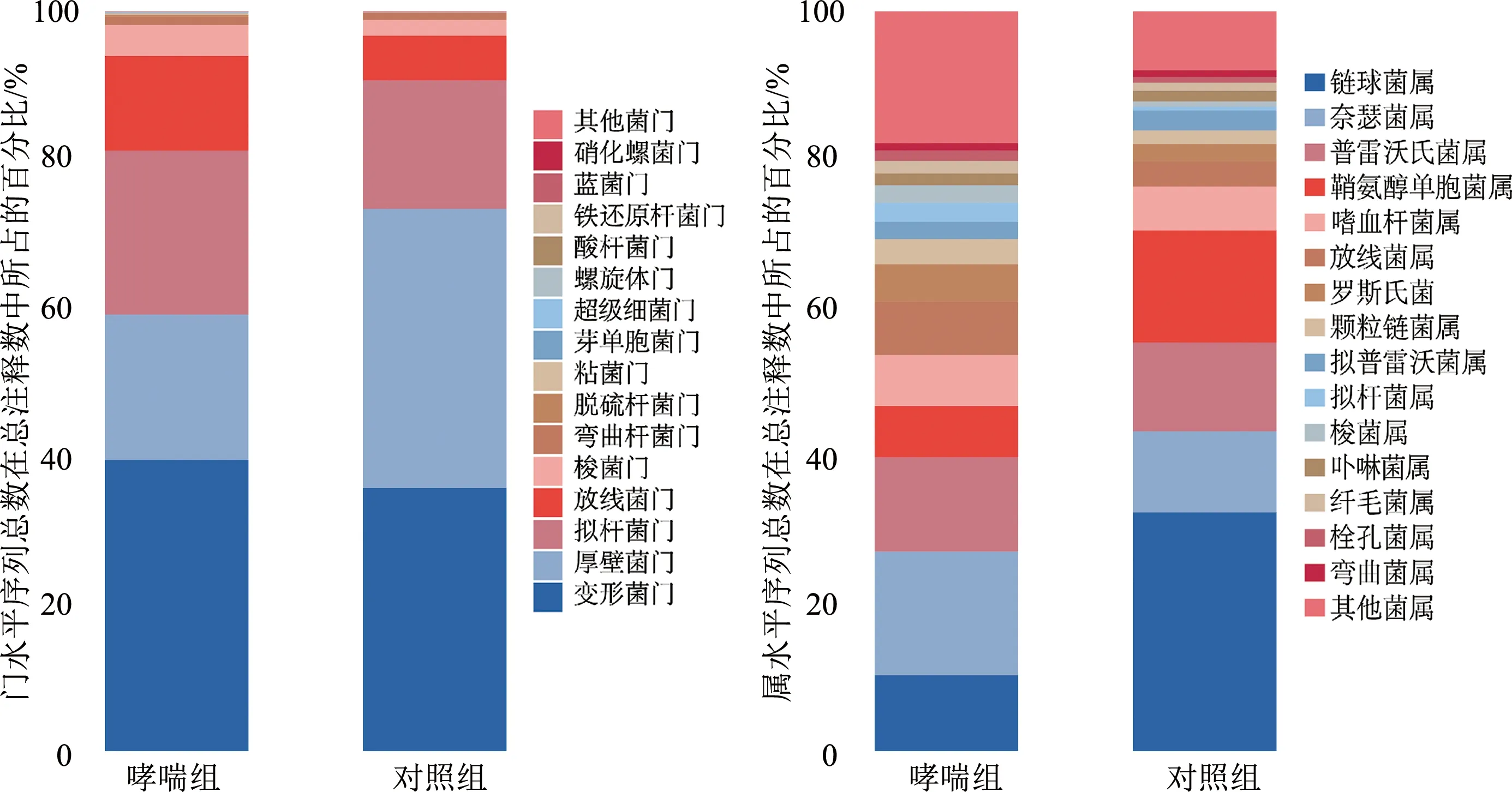
图4 门水平和属水平物种丰度累积柱状图
在属水平,对相对丰度排在前15的菌属进行分析,结果表明链球菌属、奈瑟菌属、普雷沃氏菌属、鞘氨醇单胞菌属、嗜血杆菌属、放线菌属为哮喘组和对照组共有优势菌属。与对照组相比,哮喘组中奈瑟菌属的相对丰度显著增加(P<0.05),链球菌属、鞘氨醇单胞菌属的相对丰度明显减少(P<0.05)。
2.7 差异物种分析
利用线性判别分析(LEfSe)进行比较,确定哮喘组与对照组间重要差异菌群。由图5A可知,哮喘组中伯克氏菌科、罗尔斯通氏菌科、颤螺菌属及乳酸杆菌科等显著高丰度富集。对照组中链球菌、杆菌、乳酸菌及厚壁菌门等显著高丰度富集。由图5B可知,哮喘组中起到重要作用的微生物类群有高温单孢菌科、鞘脂单孢菌科及鞘脂单孢菌目等;对照组中起到重要作用的微生物类群有纤细芽孢杆菌纲、韦荣氏球菌科、德沃斯氏菌科等。
3 讨论
哮喘是儿童期最常见的慢性呼吸道疾病,其患病率呈逐渐上升趋势。哮喘发病机制复杂,与遗传、呼吸道感染、异常的免疫反应等有关,其中呼吸道微生物群对哮喘的发生、发展起着重要作用[9]。本研究选取学龄前儿童为研究对象,比较哮喘组和对照组两组间呼吸道菌群的差异。结果发现,哮喘组呼吸道菌群多样性和丰富度明显升高。
本研究结果显示,在门水平上,哮喘组和对照组中的共有优势菌门主要为变形菌门、厚壁菌门、拟杆菌门、放线菌门、梭杆菌门及弯曲杆菌等,与Hufnagl等[10]的研究结果基本一致。哮喘组中变形杆菌、放线菌门、梭杆菌门、弯曲杆菌的相对丰度增加,而厚壁菌门的相对丰度明显下降。Huang等[11]研究报道,在哮喘患者中,变形菌属(包括嗜血菌属和奈瑟菌属)富集,而这些菌属与支气管高反应性相关[9]。Hilty等[5]研究结果表明,哮喘患者支气管肺泡灌洗液中变形菌属与哮喘发病密切相关。Huang等[11]研究报道,厚壁菌门在哮喘儿童中的丰度显著降低,提示厚壁菌门的细菌生态失调可能与哮喘儿童的哮喘风险增加相关。
在属水平,链球菌属、奈瑟菌属、普雷沃氏菌属、鞘氨醇单胞菌属、嗜血杆菌属及放线菌属等为哮喘组和对照组共有优势菌属。哮喘患儿中奈瑟菌属相对丰度明显增加,而链球菌属、鞘氨醇单胞菌属,相对丰度减少。有文献报道,用于益生菌的细菌主要属有乳酸菌(厚壁菌门、乳酸菌目、乳酸菌属、链球菌、肠球菌),放线菌属(双歧杆菌目、双歧杆菌属)和非致病性大肠杆菌[12]。本研究结果显示哮喘患儿上呼吸道中的有益细菌明显减少。临床研究证明肠道摄入益生菌和益生元可以降低哮喘发病率[9]。此外,荣磊等[13]研究表明,外周血调节性T细胞和Th17细胞比例的失衡与哮喘发病密切相关,而口服乳鼠李糖乳杆菌和双歧杆菌能够诱导抗原特异性调节性T细胞,有助于抑制过敏反应[9]。因此,通过调节气道菌群平衡,增加有益菌群可用于预防和控制哮喘发作。
综上所述,哮喘患儿菌群丰富度及多样性明显增加,菌群结构紊乱。呼吸道微生态的失衡与哮喘的发生发展有关,通过改善呼吸道菌群稳态的生态观,将为哮喘的治疗及预防提供新的策略。本研究结果显示哮喘组与对照组在物种多样性和丰度及物种差异等各指标之间具有差异性,由于样本量偏少,仍需扩大样本量,进一步明确哮喘患儿与健康儿童的差异菌种。
