分枝杆菌非典型错配修复及其在抗生素耐药中的研究进展
2023-11-24向莎莎谢建平
向莎莎,谢建平
综 述
分枝杆菌非典型错配修复及其在抗生素耐药中的研究进展
向莎莎,谢建平
西南大学生命科学学院现代生物医药研究所,重庆 400715
错配修复(mismatch repair, MMR)是生物体DNA复制后的一种常见修复系统,对于维持基因组稳定性至关重要,其关键步骤由MutS和MutL蛋白家族的成员执行,尽管这种修复途径十分重要,但在许多古菌和放线菌基因组中并不存在MutS或MutL的同源蛋白。这类细菌(例如分枝杆菌等)采用另一种非典型的MMR途径,由核酸内切酶EndoMS/NucS发挥关键作用,与典型MMR蛋白(MutS/MutL)相比没有结构同源性。EndoMS/NucS介导的非典型错配修复在分枝杆菌DNA修复、突变和同源重组以及抗生素耐药等方面发挥重要作用。本文通过对比典型MMR途径和非典型MMR途径,深入阐述了分枝杆菌EndoMS/NucS介导的非典型MMR途径及其最新进展,以期为分枝杆菌错配修复分子机制带来新见解以及对分枝杆菌抗生素治疗提供研究线索。
分枝杆菌;错配修复;EndoMS/NucS;抗生素耐药
外源性和内源性因素都可能会对生物体DNA产生损伤,这些损伤需要进行及时的修复以避免对细胞产生不利影响,如突变和细胞死亡等。因此,有效的DNA修复途径对于DNA损伤修复和基因组稳定性维持至关重要。原核生物和真核生物保守的DNA修复途径主要包括碱基切除修复(base excision repair,BER)、核苷酸切除修复(nucleotide excision repair,NER)、错配修复(mismatch repair,MMR)、同源重组(homologous recombination,HR)和非同源末端连接(non-homologous end joining,NHEJ)等[1]。错配修复系统是维持基因组完整性的主要途径,主要修复因DNA复制错误或含有序列差异的DNA之间的重组而导致的碱基插入/删除错配来维持基因组的稳定性。除了DNA损伤修复,MMR还参与有丝分裂和减数分裂重组、细胞凋亡、免疫球蛋白基因重排以及体细胞超突变等过程,且错配修复的缺陷会导致超变异性和微卫星不稳定等现象[2]。错配修复存在典型和非典型途径,大部分细菌和真核生物中是典型的错配修复,由MutS (siegel mutator)和MutL (mutator)蛋白来执行其修复活性,非典型途径则是古菌和放线菌中依赖EndoMS/ NucS的错配修复途径[3,4]。本文从分子机制、生化分析和遗传进化等方面对典型错配修复和非典型错配修复进行了综述,总结了分枝杆菌非典型MMR的修复过程和研究进展,并表明了其在分枝杆菌抗生素耐药中的潜在作用。
1 典型错配修复
1.1 细菌典型错配修复
生物DNA复制时可能会产生碱基配对错误,一般由聚合酶错误掺入、异等位基因亲本DNA之间的重组以及化学或物理因素损伤核苷酸引起[5]。为应对此类错误情况,生物体产生了相应的错配修复机制。它既可以修复DNA复制过程中出现的碱基错配,又可以消除由于含简单重复序列的同源序列间遗传重组出现的不配对序列,有效防止DNA复制差错的产生[6]。一般的错配修复途径是识别出正确的链,切除掉不正确链的错配部分,通过DNA聚合酶III和DNA连接酶的作用,最后合成正确配对的双链DNA。这一修复途径在细菌、真核生物中均保守存在,对维持基因组稳定性起重要作用。
大肠杆菌()是研究细菌典型MMR途径的模式生物,存在由甲基化介导的错配修复系统[7]。DNA复制过程中,模板链的GATC序列的A的N6位被DNA甲基化酶甲基化,而子链在DNA合成后,在几秒到几分钟内保持未甲基化状态,因此新合成的DNA双链分子处于半甲基化状态[8,9]。错配修复系统正是利用这一原理,以模板链的碱基为模板,对子链的错配核苷酸进行切割和修复。
高度保守的MutS同源蛋白和MutL同源蛋白是错配修复的基本组成部分[5]。典型的大肠杆菌错配修复过程是:当DNA发生错配时,MutS二聚体(识别关键氨基酸:F36和E38)识别并结合到错配碱基部位[10],然后MutS(结合关键氨基酸:K620、D693、E694、S668和T669等)结合三磷酸腺苷(ATP)并发生构象变化[11],在DNA上形成滑动钳,招募MutL二聚体蛋白特异性结合到滑动钳上形成突环复合物[12],复合体随ATP水解而在DNA上滑动,如果遇到碱基对错配引起的隆起就会停留,开始把两边的DNA双链拉扯,成环,直至遇到GATC序列。随后该复合物招募核酸内切酶MutH(hill mutator)结合到MutSL上并激活MutH的内切酶活性[13],MutH能够通过新合成的DNA中GATC位点腺嘌呤是否被甲基化酶Dam(DNA adenine methylase)酶甲基化来区分子链和亲本DNA链,从而能够根据GATC位点的位置从错配位点的5′端或3′端来切割含错配碱基的DNA链产生切口。之后,MutL激活解旋酶UvrD (MutU)[14,15],UvrD将替代MutH并将DNA解螺旋至错配位点,若切开处位于错配碱基的3′侧,则由核酸外切酶I (exonuclease 1,Exo1)或核酸外切酶X (exonuclease X,ExoX)沿3′→5′方向切除核酸链,若切开处位于错配碱基的5′侧,则由核酸外切酶VII或RecJ核酸外切酶沿5′→3′方向切除核酸链[16,17]。接着在单链DNA结合蛋白(single stranded-DNA binding protein, SSB)和DNA聚合酶III的作用下修补缺口并在DNA连接酶的作用下连接,最后由甲基化酶Dam酶甲基化子链形成成熟的DNA链[10,17]。
1.2 真核生物典型错配修复
DNA错配修复的重要性使得它在从细菌到人类的生物体中都存在。真核生物的错配修复基本上遵循相同的起始、识别、去除和再合成步骤,但与细菌相比存在一些差异或者过程更加复杂[18]。在真核生物中,MMR与DNA复制紧密相关,所以在S期发挥最高活性[19]。而MutS和MutL同源二聚体蛋白变成异源二聚体,功能也随之变化。
例如,在哺乳动物细胞中,碱基错配由两个MutS同源物之一识别,MutSα(MSH2/MSH6的异源二聚体)或MutSβ(MSH2/MSH3的异源二聚体)[20]。MutSα主要与4个核苷酸大小的碱基错配或小插入/缺失结合,而MutSβ可以与多达24个核苷酸的大插入或缺失结合[21]。其他的MutS同源物如MSH4-5、MSH1-3参与减数分裂等过程[22]。在切除步骤中,MutSα或MutSβ募集异二聚体MutLα (人类中是MLH1/PMS2,在酵母中是MLH1/PMS1),形成四聚体复合物[23]。真核细胞不表达类似于大肠杆菌MutH的蛋白质,真核MutLα具有潜在的核酸内切酶活性,由增殖细胞核抗原(proliferating cell nuclear antigen,PCNA)、复制因子C (replication factor C,RFC)和MutSα等相关因子激活[24]。在DNA切割步骤后,具有5′→3′ DNA核酸外切酶活性以及5′端内切酶活性的核酸外切酶1,被MSH2和MLH1招募并激活[25,26],激活的Exo1切除新合成的DNA链,DNA切除间隙被DNA聚合酶δ填充。当DNA再合成完成时,最后剩余的缺口被DNA连接酶连接[27],完成修复。其中MutS和MutL是ATP酶,它们在真核细胞中发挥作用依赖ATP,在MutSα识别和结合错配的过程以及在MutLα切割链和沿DNA转移等的过程中,都需要ATP的水解[18,28]。
MMR系统巧妙的分子机制和高效的修复能力维持了生物体基因组的稳定性。虽然MMR的一般步骤、分子机器的作用过程和机制被发现和理解,但其中更复杂的调控网络还不清楚,还需要继续探索研究。
2 分枝杆菌非典型错配修复
分枝杆菌是一组多样性的细菌,包括相关的人类病原体,如导致结核病的结核分枝杆菌()。在感染过程中,结核分枝杆菌会暴露在一系列的环境损伤中,包括宿主的免疫反应,同时也会暴露于多种类型的DNA损伤剂中[4]。像所有细胞生物一样,它们也具有有效的DNA修复系统,这些系统在分枝杆菌发病机制中起着至关重要的作用。然而,分枝杆菌基因组中未发现典型MMR系统[29],而是拥有另一种非典型的错配修复系统,其主要参与蛋白是DNA修复蛋白EndoMS/NucS,与典型MMR关键因子MutS和MutL无结构同源性[30],是一种错配特异性内切酶,在分枝杆菌体内有避免突变发生和抗重组的作用,这是典型MMR的两个标志[31](表1)。
2.1 EndoMS/NucS蛋白结构和修复机制
NucS是一种DNA修复核酸内切酶,最初被鉴定为一种新型古菌核酸酶,它是在筛选DNA滑动钳结合蛋白时分离出来的,并表征为结构特异性的DNA核酸内切酶[32]。在火球菌属文库中发现其具有错配碱基特异性内切酶活性,因此将其重命名为EndoMS(mismatch-specific endonuclease)[33]。虽然EndoMS最初是在古菌中发现的,但已经表明,这种DNA修复蛋白广泛分布在那些缺乏典型MMR蛋白MutS和MutL的物种中,包括放线菌[34]。
EndoMS/NucS是放线菌和古菌中高度保守的DNA修复蛋白。通过EndoMS/NucS家族代表性成员之间的序列比对显示出它们都保留了高度的序列相似性。分析来自分枝杆菌中耻垢分枝杆菌(Mycobacterium smegmatis MC-155)和结核分枝杆菌等的NucS序列,发现它们包含一个N端DNA结合区,预测为DNA底物的识别位点,其次是负责DNA切割的C端催化区,具有核酸酶活性(图1A)[31]。
在古菌中,EndoMS折叠成双结构域,N端DNA结合域通过短连接器连接到C端催化结构域。该酶是一种二聚体,两个N端结合结构域被一个疏水核心附着和两个C端催化结构域在两侧不对称排列形成C1N2N1C2的结构,这种折叠在放线菌中也保守[34,35]。晶体结构还表明,N末端DNA结合域折叠成半封闭β桶结构,由两层反平行片组成(八条β链和一条α螺旋),该区域参与酶二聚化,用来维持蛋白质折叠和稳定。结构比较表明,EndoMS N末端结构域具有独特的折叠,与ssDNA结合蛋白有一定的相似[30]。C端催化域包含一个α/β折叠,由五股中央β片和四个侧翼α螺旋组成,类似一个短的核酸内切酶折叠,保留了RecB样核酸酶重要基序中的保守活性位点[34,36],这些活性位点在分枝杆菌中也存在(图1B)。
由EndoMS/NucS和MutS-MutL物种分布可知,EndoMS/NucS只存在于370种生物(60种古菌和310种细菌)中,而在真核生物和病毒中不存在。EndoMS/NucS的系统发育图谱显示该蛋白质呈现出分散分布的特点。有趣的是,一些细菌中EndoMS和MutS-MutL共存,而一些缺乏EndoMS的细菌中也缺乏MutS-MutL。对完整的EndoMS以及N端和C端区域序列和系统发育独立分析可知,EndoMS具有古菌起源,并作为两个独立的蛋白质结构域的组合而出现。N末端和C末端区域都可能出现在古菌谱系中。C末端区域通过水平基因转移(horizontal gene transfer, HGT)到极少数真核生物和一些细菌中,在这些细菌中,C末端结构域与EndoMS之外的其他区域结合。而在古菌中,N末端区域和C末端区域融合产生了完整的EndoMS。EndoMS在许多古菌中扩增,但也在一些古菌中消失。完整的EndoMS 蛋白至少通过两次独立的HGT事件转移到了细菌中,一次是转移到了一些热球菌属物种中,另一次是转移到了放线菌中[31]。
对EndoMS酶活性分析表明,EndoMS是一种结构特异性核酸内切酶,能够识别和切割错配的DNA底物。该蛋白以二聚体的形式特异性结合五种错配方式(G/T,G/G,T/T,T/C,A/G),并在错配碱基两侧的第三个磷酸二酯键对称地切割两条链,形成可被连接酶连接的5′端突出的双链断裂[33,37]。大肠杆菌中,DNA复制的保真度由三个主要过程决定:DNA聚合酶的核苷酸插入保真度、相关的3′-5′校对外切酶去除错误插入的核苷酸和复制后的错配修复[38,39]。DNA复制校对由基因编码的3′-5′外切酶ε执行。ε-外切酶亚基与DNA聚合酶PolIIIα反式结合,并在DNA复制过程中切除错误插入的碱基。结核分枝杆菌中DNA复制的保真度过程相似,但结核分枝杆菌DnaQ不能与结核杆菌DnaE1聚合酶稳定结合。分析发现,分枝杆菌复制聚合酶DnaE1编码了一种新的编辑功能,通过其PHP结构域(polymerase and histidinol phosphatase domain)的3′-5′外切酶活性对DNA复制进行校对,不需要DnaQ的结合。逃避了以上校正过程的错配由NucS介导的错配修复进行修复(图2)[40]。
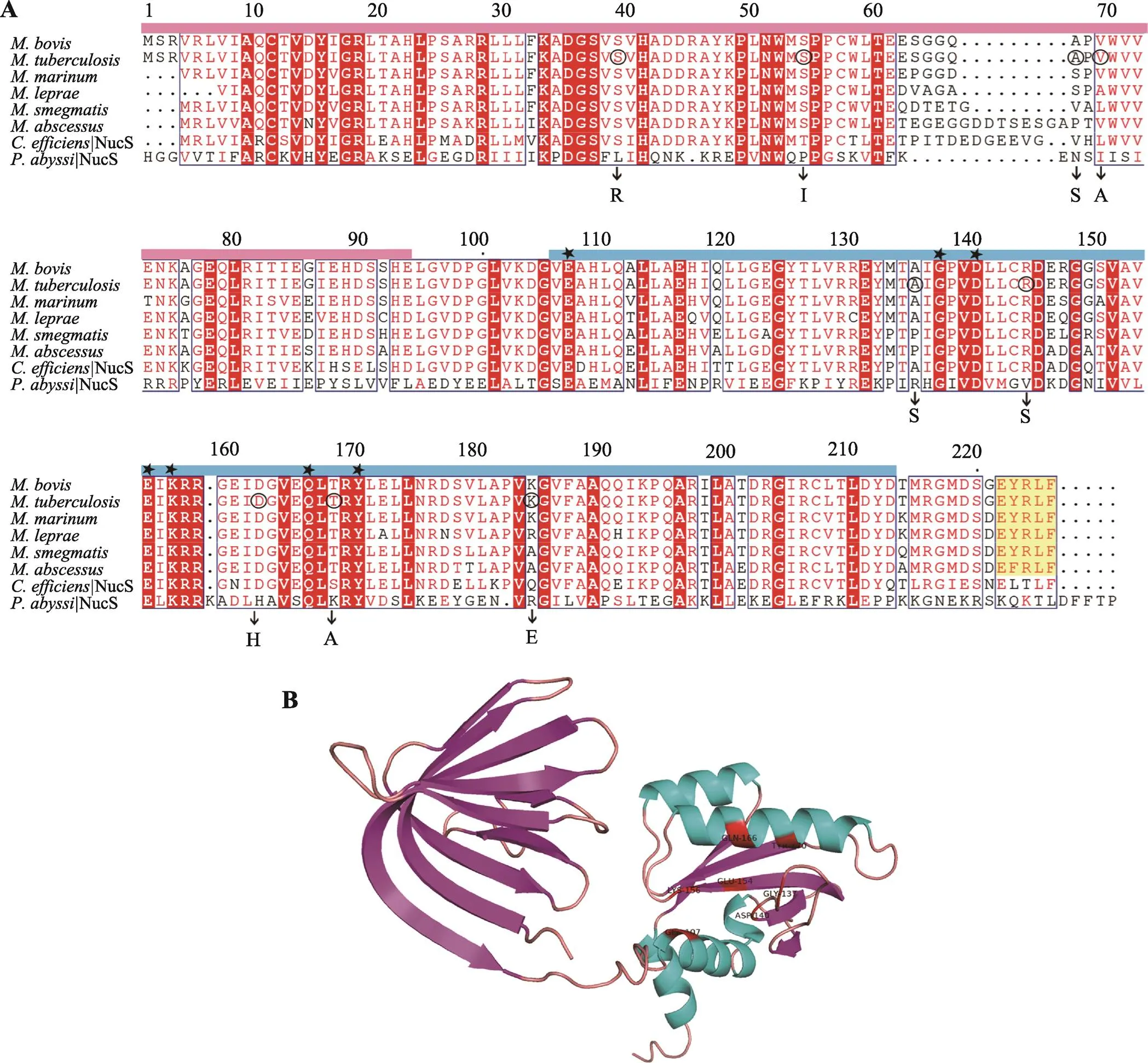
图1 EndoMS/NucS序列多序列比对及蛋白晶体图
A:分枝杆菌及古菌中的EndoMS/NucS氨基酸序列比对。粉色区域为N端结构域,蓝色区域为C端区域,黄色区域为β钳结合序列。核酸酶活性所需的催化残基用黑色星形标出,圈出的氨基酸为临床结核分枝杆菌错义单核苷酸多态性位点,箭头下为突变后的氨基酸;B:结核分枝杆菌H37Rv的NucS蛋白质单体晶体结构(Uniprot:P9WIY5)。红色标记是A中标出的氨基酸C端核酸酶活性位点。
在古菌和细菌中,EndoMS的切割活性需要滑动钳的帮助,古菌中的滑动夹是PCNA,与EndoMS C端的PIP基序(PCNA-interacting peptide motif)相互作用,PCNA将DNA聚合酶的催化亚基和其他蛋白质拴在DNA上参与复制过程,可增强核酸内切酶EndoMS的切割活性[41,42]。PIP基序在放线菌中保守存在,由五个氨基酸残基([E/K]-[L/Y]-[T/R]-L-F)组成[37](图1A)。在放线菌中除了PCNA滑动钳外还有如谷氨酸棒状杆菌编码的DNA聚合酶III的β亚基(PCNA的同源蛋白),以及结核分枝杆菌DnaE1聚合酶的β亚基等滑动钳[34,43]。此外,还发现在古菌的热球菌属中,与共转录,后者编码古菌 HR关键蛋白。的细菌同源物是,但在细菌中,RadA或RecA是否参与了NucS依赖性活动所产生的DSB的校正还有待研究[33]。
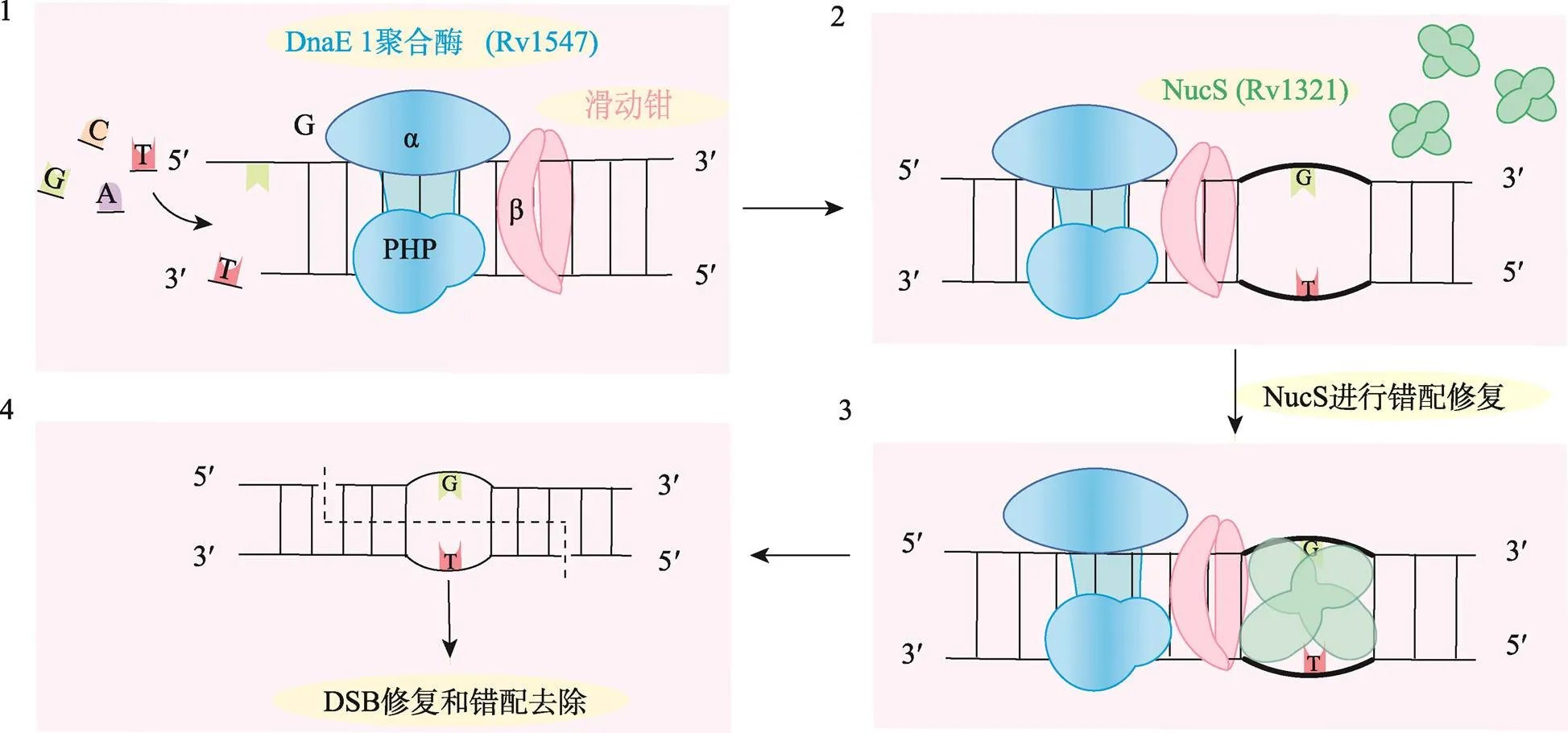
图2 结核分枝杆菌的非典型MMR途径模型
1:复制过程中Dna E1聚合酶进行碱基选择,并通过其PHP结构域进行校对活动(3′-5′核酸外切酶)。2:NucS对产生的错配进行识别和修复。3:NucS与含有错配的ds DNA结合,通过与滑动夹相互作用激发其活性。4.:NucS在错配附近切割两条链,形成双链断裂。最后,DSB和错配可以通过HR途径或其他DSB修复机制进行修复。DnaE1聚合酶(α亚基,蓝色)、滑动夹(β亚基,粉色)和NucS二聚体(绿色)。Rv1547:DNA聚合酶III,一种多链酶,负责复制合成,该DNA聚合酶还具有3′-5′外切核酸酶活性。Rv1321:NucS,一种DNA修复内切核酸酶。
2.2 EndoMS/NucS蛋白的生物学功能
Castañeda-García等[31]通过筛选耻垢分枝杆菌转座子突变库,发现耻垢分枝杆菌中的()基因缺失后,产生显著的超变异表型。衍生型菌株与野生型菌株相比,自发出现的利福平和链霉素耐药性分别增加了150倍和86倍,在其他放线菌物种的的缺失也导致菌株对利福平和链霉素突变率增加。此外,通过突变积累实验(mutation accumulation experiments, MA)发现的缺失导致耻垢分枝杆菌全基因组的总突变率增加了31倍[44]。说明NucS在避免突变方面起重要作用[37]。
DNA复制过程中的错误,转换是最常见的碱基对替换(base pair substitutions,BPSs)突变,而转换偏向突变谱是典型MMR系统的一个特征[45]。为了表征其突变谱,对耻垢分枝杆菌和谷氨酸梭菌()等菌株野生型和菌株的靶基因()进行测序,分析对利福平产生耐药性的突变类型。发现菌株表现出大量转换突变的独特积累,且菌株中检测到的所有突变都是特定的转换(A : T→G : C或G : C→A : T),说明NucS可能通过参与控制基因组G : C含量的增加来防止转换突变的积累。通过整合报告质粒分析BPSs和插入缺失发现缺失后1-bp插入或缺失以及颠换的修复率非常低,说明NucS对短插入缺失和颠换缺乏修复活性,这是分枝杆菌MMR系统的独特特征[31,44]。
MMR有助于控制基因组保真度,不仅避免产生点突变,而且防止同源重组的发生[46,47]。因此,MMR基因的失活不仅导致高突变表型,而且增加了同源重组率[48]。在缺乏的情况下,非相同序列之间的重组率显著增加,在95%的同源性下,重组率提高了3倍,在90%的同源性下,重组率提高了10倍[31]。MMR级联参与抑制同源重组已被广泛证实是MMR系统的显著特征之一,NucS调控同源重组的作用更加将其确定为非典型MMR系统的核心[49,50]。
3 分枝杆菌中错配修复与抗生素耐药性
在细菌中,突变率增加的等位基因(突变体或超突变体)在自然界中的分离频率变高[51],由于这些等位基因增加了有利突变的可能性,它们的出现可能会产生抗生素抗性[34,52]。细菌的超变异性通常与MMR成分的缺陷有关。作为抗生素激发的响应,MMR活性丧失也可能引起短暂性超突变[53]。超变异性可能不仅对抗生素耐药性有重要影响,而且对病原体的毒力和传播性等也有重要影响[54]。
结核分枝杆菌具有克隆群体结构,菌株之间几乎没有任何持续的DNA转移[55]。它仅通过突变获得抗生素耐药性,并且菌株之间的突变率存在差异[56]。因此,高可变变体(hypermutable variants)的选择可能在这种病原体中特别有利。通过缺失来寻找结核分枝杆菌超突变临床分离株,对数据库中可用的1600个临床结核分枝杆菌菌株的nucS序列的分析发现了9个错义单核苷酸多态性(single nucleotide polymorphisms, SNPs)(图1A)。将9个多态性位点加上野生型整合到耻垢分枝杆菌Δ染色体上来检测对NucS活性的影响,结果发现5个等位基因显著增加突变率,其中等位基因S39R表现出最强的突变表型(83倍)[31],说明存在受NucS调节的高突变结核分枝杆菌临床株。
为了解决非典型MMR的失活是否会通过突变增加耐药性的获得,Fressatti等[57]构建了一种菌株,并在病原体脓肿分枝杆菌()中进行探索。结果发现的缺失导致突变表型产生,使得脓肿分枝杆菌对大环内酯类和氨基糖苷类药物以及氟喹诺酮类等药物[58]的耐药性增加。与野生型菌株相比,非典型MMR的失活使脓肿分枝杆菌对大环内酯类克拉霉素和氨基糖苷类阿米卡星、庆大霉素和阿布拉霉素耐药的自发突变体增加了10~22倍。23S rRNA基因和16S rRNA基因测序结果显示菌株对大环内酯类和氨基糖苷类药物的耐药性与靶基因突变有关,并可能由缺乏驱动。此外,在分析克拉霉素耐药性的获得时,与野生型菌株相比,在脓肿分枝杆菌菌株中检测到不同的突变谱。这些结果都说明脓肿分枝杆菌需要NucS依赖性非规范MMR途径来防止通过突变产生耐药菌株。
此外,还有研究发现,在一些药物的处理下结核分枝杆菌中的表达也显著下降,如维拉帕米Verapamil的处理下,表达差异值在–2.93左右,在羰基氰化物间氯苯腙CCCP处理下,差异值在–2.59以及在2-硝基苯甲酸DNTB处理下,差异值在–2.37左右等[59]。
4 结语与展望
迄今为止,错配碱基的修复被认为是一种高度保守的机制,其关键步骤由MutS和MutL蛋白家族的成员执行。但几乎所有放线菌和许多古菌成员都缺乏MutS或MutL同源物,它们存在一种错配特异性核酸内切酶EndoMS/NucS,能够在体外识别和切割dsDNA中不匹配的碱基。现在EndoMS/NucS已被确定为非典型错配修复途径的关键蛋白质,作用于分枝杆菌碱基转换等修复,对于维持基因组稳定性至关重要。
NucS的缺失导致分枝杆菌超突变体表型、转换偏向突变谱的产生以及同源重组的增加。在结核分枝杆菌的临床分离株中,也已经检测到可能对突变率产生影响的多态性NucS突变体。更重要的是,NucS缺陷突变体在分枝杆菌抗生素耐药性的获得中也发挥重要作用。但EndoMS/NucS介导的非典型MMR途径还仍有许多疑问未解决,要充分描述非典型MMR,仍有许多关于相关蛋白活性机制、调控和表达、突变效应以及进化后果和临床方面的问题有待回答,如非典型MMR途径对不同损伤的修复能力?参与非典型MMR途径的重要成分还有哪些?调控EndoMS/NucS表达的因素或分子有哪些?在同时存在EndoMS/NucS和MutS-MutL的生物中如何选择两种方式之一或者都使用?分枝杆菌中非典型MMR途径与HR途径或其他双链断裂修复途径的关系?NucS缺陷突变体对分枝杆菌抗生素联用的作用以及与其他临床超突变的关系等,还需要人们进一步的探索。深入了解分枝杆菌NucS介导的非典型MMR途径有助于对分枝杆菌修复机制进行更深一步认识以及揭示克服分枝杆菌耐药性的新策略。
[1] Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis., 2017, 58(5): 235–263.
[2] Olave MC, Graham RP. Mismatch repair deficiency: the what, how and why it is important., 2022, 61(6): 314–321.
[3] Lenhart JS, Pillon MC, Guarné A, Biteen JS, Simmons LA. Mismatch repair in Gram-positive bacteria., 2016, 167(1): 4–12.
[4] Dos Vultos T, Mestre O, Tonjum T, Gicquel B. DNA repair inrevisited., 2009, 33(3): 471–487.
[5] Fishel R. Mismatch repair., 2015, 290(44): 26395–26403.
[6] Hsieh P, Yamane K. DNA mismatch repair: molecular mechanism, cancer, and ageing., 2008, 129(7–8): 391–407.
[7] Putnam CD. Evolution of the methyl directed mismatch repair system in., 2016, 38: 32–41.
[8] Barras F, Marinus MG. The great GATC: DNA methylation in., 1989, 5(5): 139–43.
[9] Hu CK, Zhao YQ, Sun HY, Yang YX. Synergism of Dam, MutH, and MutS in methylation-directed mismatch repair in Escherichia coli., 2017, 795: 31–33.
[10] Groothuizen FS, Sixma TK. The conserved molecular machinery in DNA mismatch repair enzyme structures., 2016, 38: 14–23.
[11] Junop MS, Yang W, Funchain P, Clendenin W, Miller JH. In vitro and in vivo studies of MutS, MutL and MutH mutants: correlation of mismatch repair and DNA recombination., 2003, 2(4): 387–405.
[12] Jeong C, Cho WK, Song KM, Cook C, Yoon TY, Ban C, Fishel R, Lee JB. MutS switches between two fundamentally distinct clamps during mismatch repair., 2011, 18(3): 379–385.
[13] Au KG, Welsh K, Modrich P. Initiation of methyl-directed mismatch repair., 1992, 267(17): 12142–12148.
[14] Yamaguchi M, Dao V, Modrich P. MutS and MutL activate DNA helicase II in a mismatch-dependent manner., 1998, 273(15): 9197–9201.
[15] Grilley M, Griffith J, Modrich P. Bidirectional excision in methyl-directed mismatch repair., 1993, 268(16): 11830–11837.
[16] Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology., 1996, 65: 101–133.
[17] Hsieh P. Molecular mechanisms of DNA mismatch repair., 2001, 486(2): 71–87.
[18] Liu DK, Keijzers G, Rasmussen LJ. DNA mismatch repair and its many roles in eukaryotic cells., 2017, 773: 174–187.
[19] Schroering AG, Edelbrock MA, Richards TJ, Williams KJ. The cell cycle and DNA mismatch repair., 2007, 313(2): 292–304.
[20] Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms., 2006, 106(2): 302–323.
[21] McCulloch SD, Gu LY, Li GM. Bi-directional processing of DNA loops by mismatch repair-dependent and -independent pathways in human cells., 2003, 278(6): 3891–3896.
[22] Manhart CM, Alani E. Roles for mismatch repair family proteins in promoting meiotic crossing over., 2016, 38: 84–93.
[23] Plotz G, Raedle J, Brieger A, Trojan J, Zeuzem S. hMutSalpha forms an ATP-dependent complex with hMutLalpha and hMutLbeta on DNA., 2002, 30(3): 711–718.
[24] Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair., 2006, 126(2): 297–308.
[25] Tran PT, Erdeniz N, Symington LS, Liskay RM. EXO1-A multi-tasking eukaryotic nuclease., 2004, 3(12): 1549–1559.
[26] Nielsen FC, Jäger AC, Lützen A, Bundgaard JR, Rasmussen LJ. Characterization of human exonuclease 1 in complex with mismatch repair proteins, subcellular localization and association with PCNA., 2004, 23(7): 1457–1468.
[27] Longley MJ, Pierce AJ, Modrich P. DNA polymerase delta is required for human mismatch repair in vitro., 1997, 272(16): 10917–10921.
[28] Ban C, Junop M, Yang W. Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair., 1999, 97(1): 85–97.
[29] Mizrahi V, Andersen SJ. DNA repair in. What have we learnt from the genome sequence?., 1998, 29(6): 1331–1339.
[30] Ren B, Kühn J, Meslet-Cladiere L, Briffotaux J, Norais C, Lavigne R, Flament D, Ladenstein R, Myllykallio H. Structure and function of a novel endonuclease acting on branched DNA substrates., 2009, 28(16): 2479–2489.
[31] Castañeda-García A, Prieto AI, Rodríguez-Beltrán J, Alonso N, Cantillon D, Costas C, Pérez-Lago L, Zegeye ED, Herranz M, Plociński P, Tonjum T, García de Viedma D, Paget M, Waddell SJ, Rojas AM, Doherty AJ, Blázquez J. A non-canonical mismatch repair pathway in prokaryotes., 2017, 8: 14246.
[32] Meslet-Cladiére L, Norais C, Kuhn J, Briffotaux J, Sloostra JW, Ferrari E, Hübscher U, Flament D, Myllykallio H. A novel proteomic approach identifies new interaction partners for proliferating cell nuclear antigen., 2007, 372(5): 1137–1148.
[33] Ishino S, Nishi Y, Oda S, Uemori T, Sagara T, Takatsu N, Yamagami T, Shirai T, Ishino Y. Identification of a mismatch-specific endonuclease in hyperthermophilic Archaea., 2016, 44(7): 2977–2986.
[34] Cebrián-Sastre E, Martín-Blecua I, Gullón S, Blázquez J, Castañeda-García A. Control of genome stability by EndoMS/NucS-mediated non-canonical mismatch repair., 2021, 10(6): 1314.
[35] Nakae S, Hijikata A, Tsuji T, Yonezawa K, Kouyama KI, Mayanagi K, Ishino S, Ishino Y, Shirai T. Structure of the EndoMS-DNA complex as mismatch restriction endonuclease., 2016, 24(11): 1960–1971.
[36] Pingoud A, Fuxreiter M, Pingoud V, Wende W. Type II restriction endonucleases: structure and mechanism., 2005, 62(6): 685–707.
[37] Takemoto N, Numata I, Su'etsugu M, Miyoshi-Akiyama T. Bacterial EndoMS/NucS acts as a clamp-mediated mismatch endonuclease to prevent asymmetric accumulation of replication errors., 2018, 46(12): 6152–6165.
[38] Kunkel TA, Bebenek K. DNA replication fidelity., 2000, 69: 497–529.
[39] Xie ZH. The fidelity mechanism of DNA synthesis., 2012, 34(6): 679–686.谢兆辉. DNA合成的忠实性机制. 遗传, 2012, 34(6): 679–686.
[40] Rock JM, Lang UF, Chase MR, Ford CB, Gerrick ER, Gawande R, Coscolla M, Gagneux S, Fortune SM, Lamers MH. DNA replication fidelity in Mycobacterium tuberculosis is mediated by an ancestral prokaryotic proofreader., 2015, 47(6): 677–681.
[41] Grabowski B, Kelman Z. Archeal DNA replication: eukaryal proteins in a bacterial context., 2003, 57: 487–516.
[42] Creze C, Ligabue A, Laurent S, Lestini R, Laptenok SP, Khun J, Vos MH, Czjzek M, Myllykallio H, Flament D. Modulation of the Pyrococcus abyssi NucS endonuclease activity by replication clamp at functional and structural levels., 2012, 287(19): 15648–15660.
[43] Ishino S, Skouloubris S, Kudo H, l'Hermitte-Stead C, Es-Sadik A, Lambry JC, Ishino Y, Myllykallio H. Activation of the mismatch-specific endonuclease EndoMS/NucS by the replication clamp is required for high fidelity DNA replication., 2018, 46(12): 6206–6217.
[44] Castañeda-García A, Martín-Blecua I, Cebrián-Sastre E, Chiner-Oms A, Torres-Puente M, Comas I, Blázquez J. Specificity and mutagenesis bias of the mycobacterial alternative mismatch repair analyzed by mutation accumulation studies., 2020, 6(7): eaay4453.
[45] Garibyan L, Huang T, Kim M, Wolff E, Nguyen A, Nguyen T, Diep A, Hu K, Iverson A, Yang H, Miller JH. Use of the rpoB gene to determine the specificity of base substitution mutations on the Escherichia coli chromosome.,2003, 2(5): 593–608.
[46] Schofield MJ, Hsieh P. DNA mismatch repair: molecular mechanisms and biological function., 2003, 57: 579–608.
[47] Tham KC, Kanaar R, Lebbink JHG. Mismatch repair and homeologous recombination., 2016, 38: 75–83.
[48] Tham KC, Hermans N, Winterwerp HHK, Cox MM, Wyman C, Kanaar R, Lebbink JHG. Mismatch repair inhibits homeologous recombination via coordinated directional unwinding of trapped DNA structures., 2013, 51(3): 326–337.
[49] Worth L Jr, Clark S, Radman M, Modrich P. Mismatch repair proteins MutS and MutL inhibit RecA-catalyzed strand transfer between diverged DNAs., 1994, 91(8): 3238–3241.
[50] Spies M, Fishel R. Mismatch repair during homologous and homeologous recombination., 2015, 7(3): a022657.
[51] Matic I, Radman M, Taddei F, Picard B, Doit C, Bingen E, Denamur E, Elion J. Highly variable mutation rates in commensal and pathogenic Escherichia coli., 1997, 277(5333): 1833–1834.
[52] Taddei F, Radman M, Maynard-Smith J, Toupance B, Gouyon PH, Godelle B. Role of mutator alleles in adaptive evolution., 1997, 387(6634): 700–702.
[53] Gutierrez A, Laureti L, Crussard S, Abida H, Rodríguez- Rojas A, Blázquez J, Baharoglu Z, Mazel D, Darfeuille F, Vogel J, Matic I. β-Lactam antibiotics promote bacterial mutagenesis via an RpoS-mediated reduction in replication fidelity., 2013, 4: 1610.
[54] Oliver A, Mena A. Bacterial hypermutation in cystic fibrosis, not only for antibiotic resistance., 2010, 16(7): 798–808.
[55] Gagneux S. Ecology and evolution of Mycobacterium tuberculosis., 2018, 16(4): 202–213.
[56] Ford CB, Shah RR, Maeda MK, Gagneux S, Murray MB, Cohen T, Johnston JC, Gardy J, Lipsitch M, Fortune SM. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis., 2013, 45(7): 784–790.
[57] Fressatti Cardoso R, Martín-Blecua I, Pietrowski Baldin V, Meneguello JE, Valverde JR, Blázquez J, Castañeda-García A. Noncanonical mismatch repair protein NucS modulates the emergence of antibiotic resistance in Mycobacterium abscessus., 2022, 10(6): e0222822.
[58] Daley CL, Iaccarino JM, Lange C, Cambau E, Wallace RJ Jr, Andrejak C, Böttger EC, Brozek J, Griffith DE, Guglielmetti L, Huitt GA, Knight SL, Leitman P, Marras TK, Olivier KN, Santin M, Stout JE, Tortoli E, van Ingen J, Wagner D, Winthrop KL. Treatment of nontuberculous mycobacterial pulmonary disease: an official ATS/ERS/ESCMID/IDSA clinical practice guideline., 2020, 56(1): 2000535.
[59] Boshoff HI, Myers TG, Copp BR, McNeil MR, Wilson MA, Barry CE 3rd. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action., 2004, 279(38): 40174–40184.
Progress on the non-canonical mismatch repair inand its role in antibiotic resistance
Shasha Xiang, Jianping Xie
Mismatch repair (MMR) is a common repair system after DNA replication, which is critical for maintaining genomic stability. Members of the MutS and MutL protein families are involved in key steps of mismatch repair. Despite the major importance of this repair pathway, MutS–MutL are absent in almost all Actinobacteria and many Archaea. Mycobacteria and others have another non-canonical MMR pathway, in which EndoMS/NucS plays a key role and has no structural homology compared to canonical MMR proteins (MutS/MutL). EndoMS/NucS mediated non-canonical mismatch repair plays an important role in DNA repair, mutation, homologous recombination and antibiotic resistance of. By comparing the classical and non-canonical MMR pathways, this paper reviews the EndoMS/NucS-mediated non-canonical MMR pathway inand its recent progress. We hope to bring new insights into the molecular mechanism of mycobacterial mismatch repair as well as to provide new research clues for mycobacterial antibiotic therapy.
;mismatch repair; EndoMS/NucS; antibiotic resistance
2023-09-11;
2023-10-29;
2023-11-03
国家自然科学基金项目(编号:82072246)资助[Supported by the National Natural Science Foundation of China (No. 82072246)]
向莎莎,硕士研究生,专业方向:分枝杆菌遗传学调控和分子机制。E-mail: 1977015429@qq.com
谢建平,博士,研究员,研究方向:结核分枝杆菌等重要病原致病耐药机理与新防控措施研发。E-mail: georgex@swu.edu.cn
10.16288/j.yczz.23-236
(责任编委: 张天宇)
