氟离子选择电极法测定植物性食品基质中的氟化物
2023-11-23车明秀孙学文孙嵛林
王 琳,车明秀,李 峥,孙学文,孙嵛林,刘 睿,*
(1.山东省食品药品检验研究院,山东 济南 250101;2.山东省食品药品安全检测工程技术研究中心,山东 济南 250101;3.食药环检验研究院(山东)集团有限公司,山东 济南 250100)
联合国粮食及农业组织(FAO)/国际原子能机构(IAEA)/世界卫生组织(WHO)微量元素联合专家咨询委员会,根据氟化物预防龋齿的作用,将氟列为可能的必需微量元素[1]。适量的氟摄入会促进机体骨骼的生长,预防龋齿,但如果摄入量过多又会导致氟斑牙和氟骨病[2]。食物是机体氟元素的重要摄入来源,只有了解食物中的氟含量,才能更加有效地控制氟的摄入。
氟化物广泛存在于大气、水和土壤中,若环境中的氟化物含量较高,植物生长会因长期暴露在含氟环境中对环境中的氟化物进行广泛富集,导致氟含量较高[3-4]。尤其是茶树,其作为一种富集能力很强的植物,对氟具有选择性吸收和富集能力,并且随着叶片年龄的增长,氟含量会越来越高[5]。我国将茶叶中的氟含量作为重点监测项目,因此制定了相应的食品安全国家标准,即砖茶允许含氟量≤300 mg/kg[6]。
我国的地方性氟中毒现象十分严重,而对于氟中毒目前又没有早期的监控指标,所以防止氟中毒发生的关键还在于防,即通过关注暴露的识别与控制,从源头上降低人群暴露的机会[7]。人们的日常饮食中对植物性食品基质的摄入量普遍较多,因此关注植物性食品基质中的氟含量对于控制氟的摄入量至关重要。建立和完善植物性食品基质中氟化物的检测方法,则更有利于加强对植物性食品基质中氟的监控并预防氟中毒。
目前关于食品中氟的测定方法主要包括比色法、液相色谱法、离子色谱法、气相色谱法等,这些方法各有所长,但均存在一个共同的缺点,即操作复杂、使用仪器昂贵、费时费力、灵活性差等,无法满足大批量样品的测定需求[8-9]。GB/T 5009.18—2003[10]中的氟离子选择电极法因设备操作简便、灵敏度高、选择性好、不受浊度和色度干扰等优点,在氟含量的测定上已经得到广泛应用[10]。但通过实际测定发现该标准方法存在缺陷和不足:一是方法标准曲线的线性范围设计不合理,在标准规定的线性范围内校准曲线不稳定,重复性较差,且在低浓度范围内校准曲线线性关系不好,给测定结果带来较大的误差[11-13];二是现有国家标准中方法的称样量范围较窄,对于测定氟含量低的样品时,现有称样量无法准确定量,而在测定氟含量较高的样品时,又必须对样品进行稀释后测定,对方法准确度有一定的影响[8];三是该方法未确定检出限和定量限,在实际检测过程中给检验人员的结果判定带来困扰。
为了解决目前标准(GB/T 5009.18—2003[10])中氟离子选择电极法存在的不足,本研究对标准曲线的线性范围和称样量进行了优化,同时确定了方法的定量限,并通过不同植物性食品基质进行验证,确立方法的准确性。
1 材料与方法
1.1 材料与设备
1.1.1 材料与试剂
氟标准溶液(1 000 μg/L,批号194007),国家有色金属及电子材料分析测试中心公司;柠檬酸钠、乙酸钠、高氯酸、盐酸、氟标准使用液(50.0 μg/mL)、氟标准使用液(10.0 μg/mL),国药集团药业股份有限公司;茶叶中氟成分分析标准物质(标准值319 mg/kg,编号P24307),广州谱恩科学仪器有限公司;玉米中氟成分分析标准物质(标准值15.5 mg/kg,编号RMSA030),东莞市恒准标物计量研究院有限公司。
乙酸钠溶液(3 mol/L):称取204 g 乙酸钠,溶于300 mL 蒸馏水中,加乙酸(1 mol/L)调节pH至7.0,加蒸馏水稀释至500 mL。
柠檬酸钠溶液(0.75 mol/L):称取110 g柠檬酸钠溶于300 mL蒸馏水中,加入14 mL高氯酸,再加蒸馏水稀释至500 mL。
总离子强度缓冲剂:将乙酸钠溶液(3 mol/L)与柠檬酸钠溶液(0.75 mol/L)等量混合,临用时现配制。盐酸:浓盐酸∶水(V/V)=1∶11。
1.1.2 仪器与设备
7101型氟电极1、PHS-3C型酸度计:上海雷磁仪器有限公司;perfectlON COMB F 型氟电极2、ME204T型分析天平:梅特勒-托利多公司;Hei-Tec型磁力搅拌器:德国海道夫公司;JX-2008型高速组织捣碎机:拓赫机电科技(上海)有限公司;SH-4C型磁力搅拌器:群安实验仪器有限公司。
1.2 方法
1.2.1 样品制备
样品取可食部分粉碎均匀,装入洁净密闭的容器中保存。
1.2.2 样品前处理
盐酸(1+11)提取(除茶叶外的基质):称取试样,置于50 mL 容量瓶中,加10 mL 盐酸(1+11),密闭浸泡提取1 h,提取后加25 mL 总离子强度缓冲剂,加水定容。
水煮提取(茶叶基质):称取试样,置于50 mL 具塞三角瓶中,加10 mL水,充分混匀后沸水浴15 min,取出冷却至室温,用25 mL总离子强度缓冲剂分次转移至50 mL 容量瓶中,加10 mL 盐酸(1+11),并加水定容。
1.2.3 分析步骤
定量取样:吸取提取后的上清液不少于20 mL于50 mL烧杯中(若溶液浑浊,可过滤后取滤液)。
标准曲线的制作:分别准确移取1.0、2.5、5.0、10.0 mL氟标准使用液(50.0 μg/mL)和5.0、8.0、10.0 mL氟标准使用液(10.0 μg/mL)于50 mL 容量瓶中,分别加入25 mL总离子强度缓冲剂,10 mL盐酸(1+11),并加水定容。
测定:将电极插入盛有不少于20 mL 水的50 mL塑料杯中,杯中放入磁力搅拌转子(转速保持恒定),读取平衡电位值,更换2~3次水后,待电位值平衡后,即可进行待测液与标准液的电位测定。同时做试剂空白试验。
1.2.4 结果计算
以电极电位为纵坐标,氟离子质量浓度的负对数值为横坐标,绘制标准曲线,根据试样的电位值在标准曲线上求出氟离子质量浓度。
试样中的氟含量计算公式见式(1)。
式中:X为试样中的氟含量,mg/kg;A为测定用样液中氟离子质量浓度,μg/mL;m为试样质量,g;V为样液总体积,mL。
1.2.5 数据处理
使用Microsoft Office Excel软件进行数据处理。
2 结果与分析
2.1 标准曲线线性范围的确定
原理:氟离子选择电极,电位变化规律符合能斯特(Nernst)方程式,见式(2)[10]。
式中:E为电池的电动势,V;CF-为氟离子活度,mol/L;E0为参比电极的电位(固定值),V;R为理想气体常数,8.314 J/(mol·K);F为法拉第常数,96 485 C/mol;T为绝对温度,K。
E与lgCF-成线性关系。2.303RT/F为该直线的斜率(25 ℃时为59.16)。
为了确定方法适用的线性范围,配制0.02、0.04、0.10、0.20、0.50、1.00、2.00、5.00、8.00、10.00 mg/L的系列氟标准溶液,分别采用氟电极1和氟电极2,按分析步骤对电位值进行测定,通过拟合线性方程,考察标准曲线斜率及相关系数确定标准曲线的线性范围,结果见表1。
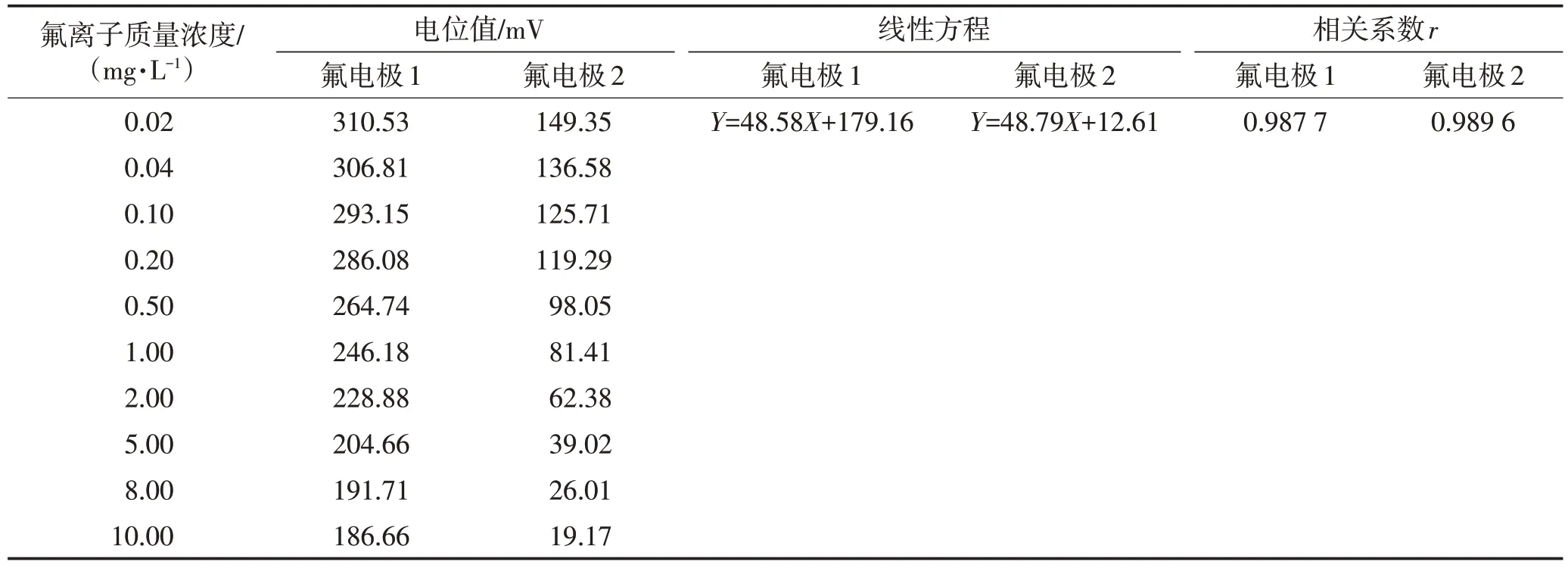
表1 0.02~10.00 mg/L线性范围的考察Table 1 Investigation on the linear range of 0.02~10.00 mg/L
由表1可见,两种品牌的电极在0.02~10.00 mg/L线性范围内,线性方程的斜率均不符合能斯特方程的理论斜率值,且r值不符合要求[14],斜率偏离理论值的原因是由于在低质量浓度范围,电位值与氟离子质量浓度的负对数值不呈现线性关系,使得标准曲线的斜率明显偏离该温度下的理论值。因此,氟离子质量浓度在0.02~10.00 mg/L 范围内氟离子选择电极法不呈线性关系。
为了确定标准曲线的适用范围,试验调整了标准曲线的起始浓度,进一步考察标准曲线的斜率和相关系数,结果见表2。

表2 线性范围的优化Table 2 Optimization of linear range
由表2可以看出,0.04~10.00 mg/L、0.10~10.00 mg/L和0.20~10.00 mg/L 氟离子质量浓度范围内,只有在0.20~10.00 mg/L范围内,两个品牌电极测定结果拟合的线性方程均符合该温度下的理论斜率值,且相关系数符合要求。因此,本试验结果证明,对于氟离子选择电极法,标准曲线的氟离子质量浓度适用范围为0.20~10.00 mg/L。
2.2 称样量的优化
为了提高方法的准确性,同时尽可能降低方法的检出限和定量限,分别对不同食品基质进行称样量的优化(通过加标回收和有证标准物质测定两种方式进行),结果见表3和表4。

表3 称样量优化(加标回收)Table 3 Sample weighing optimization(standard adding recovery)
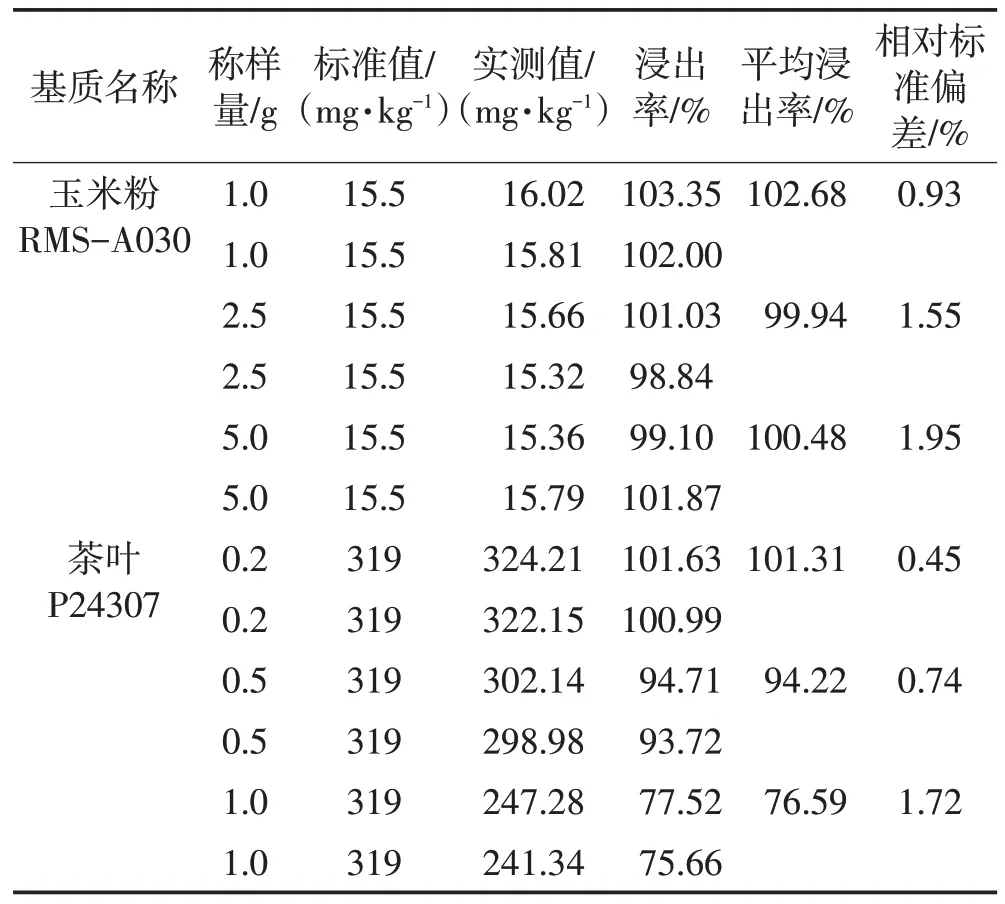
表4 称样量优化(有证标准物质)Table 4 Optimization of sample weighing(certified reference materials)
通过表3和表4可以看出,除茶叶基质外,在1.0~5.0 g 称量范围内,对于不同食品基质加标回收率或浸出率测定值均符合要求[16],但继续增大称样量会大大降低试验效率。
考虑到该法的线性浓度范围最低点为0.2 mg/L,折算到实际样品中,当称样量为1.0 g时,能准确测定样品基质中氟的最低含量为10.00 mg/kg。为了尽可能准确测定到样品中更低浓度的氟含量,通过适当提高称样量来达到降低测定低限的目的,当称样量为2.5 g 时,推算出能准确测定样品基质中氟的最低含量为4.00 mg/kg。因此,确定除茶叶外的基质的称样量适用范围为2.5~5.0 g。
对于茶叶基质,当称样量在0.2~0.5 g 范围内,浸出率均符合要求,而随着称样量的进一步增大,浸出率反而降低。原因主要有以下两点:一是由于茶叶中氟含量较高,称样量过大导致浸出不完全,从而影响测定的准确度;二是拟合的标准方程线性范围的最高值为10.0 mg/L,相当于当称样量为1.0 g 时能准确测定的茶叶基质最高氟含量为500.00 mg/kg,对于部分氟含量较高超过限量规定的不合格茶叶基质可能会超出线性范围。因此对茶叶基质的称样量范围最终确定为0.2~0.5 g。
2.3 方法检出限和定量限的确定
2.3.1 理论检出限和定量限
当校准曲线的直线部分外延的延长线与通过空白电位且平行于浓度轴的直线相交时,其交点所对应的浓度值即为该离子选择电极法的检出限[15]。通过测定10次空白电位值计算理论检出限,结果见表5。
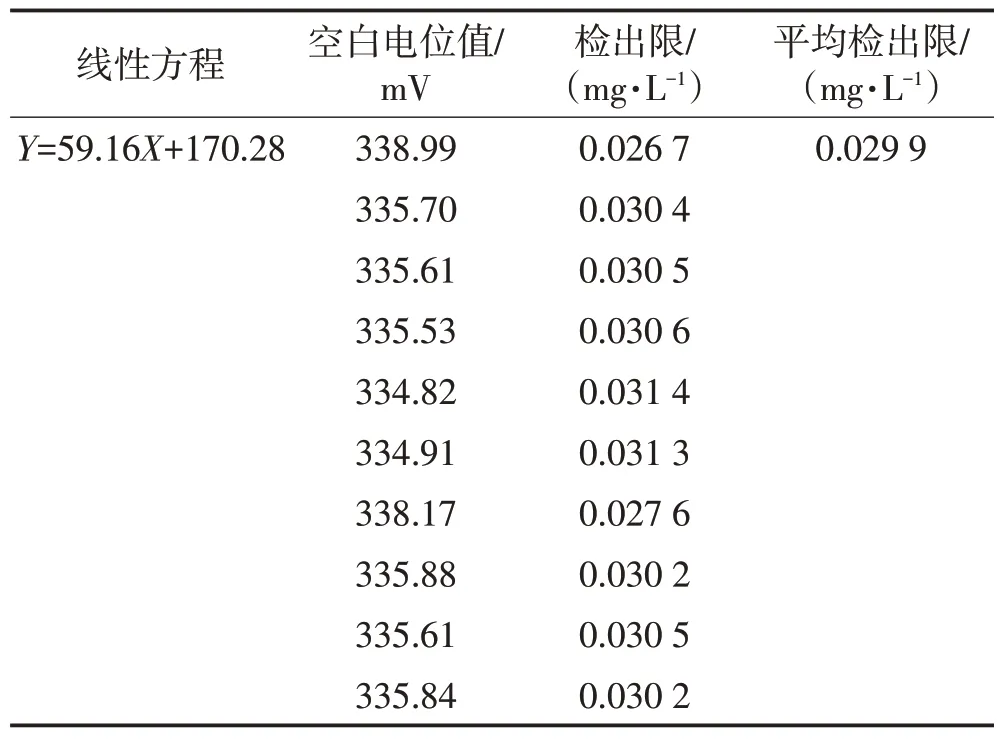
表5 理论检出限Table 5 Theoretical detection limit
由表5可以看出,理论检出限值为0.029 9 mg/L,不符合标准曲线线性范围,因此本方法不指定检出限。将标准曲线的第一个质量浓度点(0.2 mg/L)设定为理论定量限。即当称样量为2.5 g,定容体积为50 mL时,理论定量限为4.00 mg/kg;当称样量为0.2 g,定容体积为50 mL时,理论定量限为50.00 mg/kg。
2.3.2 定量限的验证
采用优化后的线性范围和称样量,分别对不同食品基质进行理论定量限的验证。验证方式为通过对不同食品基质中进行定量限水平的加标,考察加标回收率,其中韭菜和苹果的称样量均为2.5 g,茶叶的称样量为0.2 g,验证结果见表6。
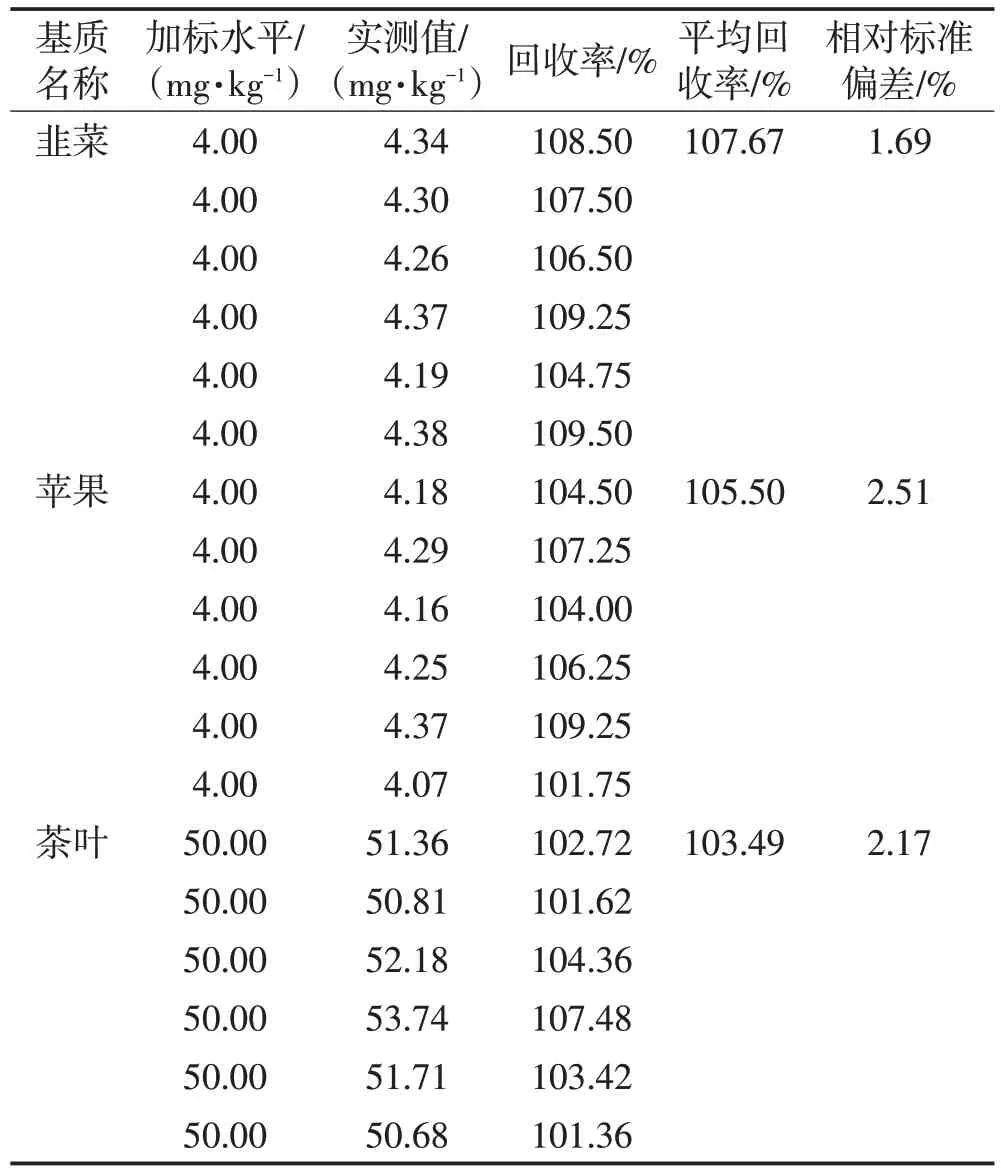
表6 定量限的验证Table 6 Validation of limit of quantification
由表6可以看出,对于不同食品基质进行定量限水平加标后的回收率均符合要求[16]。因此根据验证结果确定,对于除茶叶以外的基质,当称样量为2.5 g,定容体积为50 mL时,定量限为4.00 mg/kg;对于茶叶基质,当称样量为0.2 g,定容体积为50 mL时,定量限为50.00 mg/kg。
2.4 方法准确度和精密度的验证
选取代表性样品基质,除茶叶外的基质(韭菜、苹果)称样量均为2.5 g,分别进行定量限水平、2倍定量限水平和10 倍定量限水平的加标试验;茶叶基质称样量为0.2 g,进行本底值水平加标、1/2限量水平加标和限量水平加标,以回收率和相对标准偏差评价方法的准确度和精密度,结果见表7。
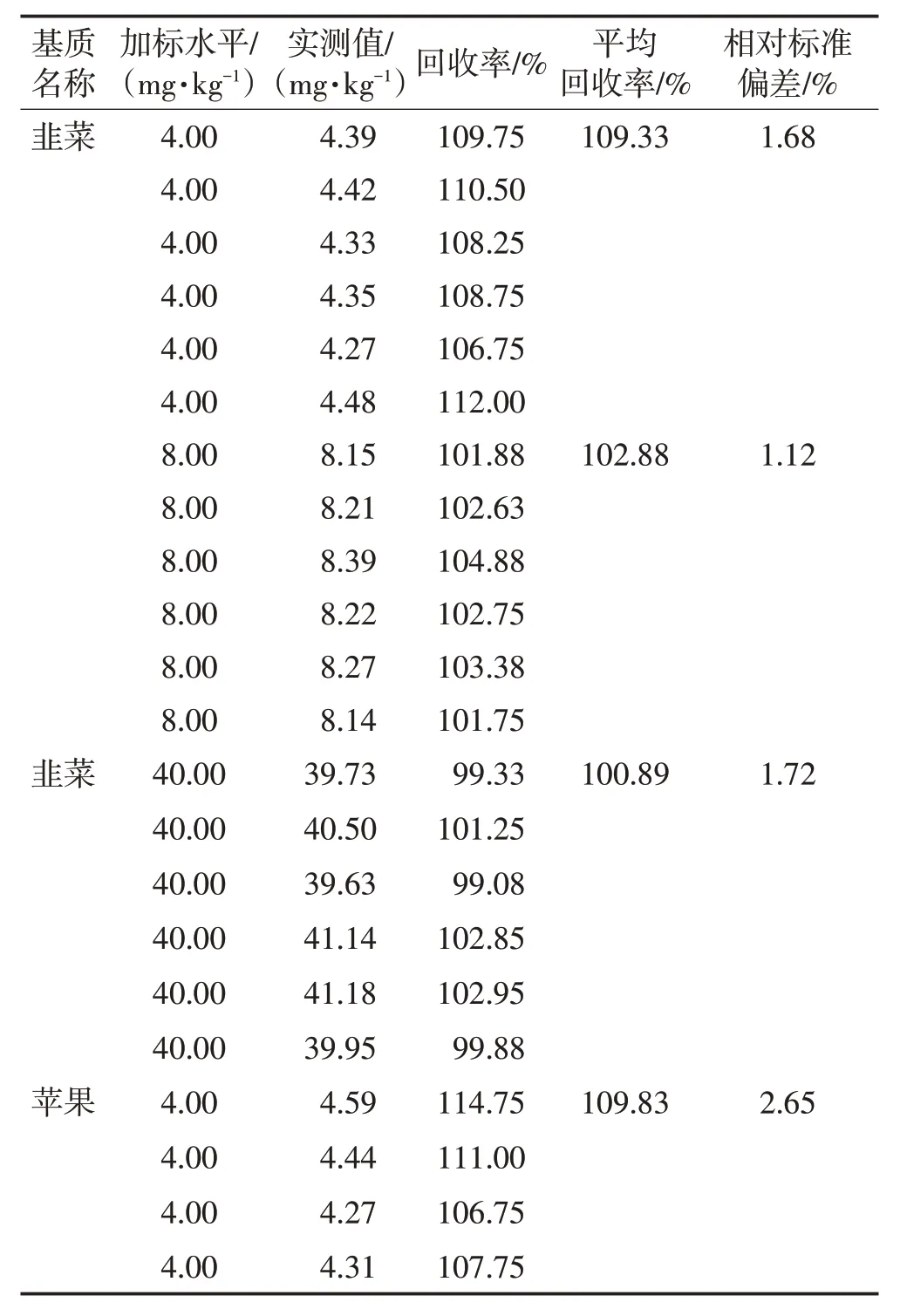
表7 准确度和精密度Table 7 Accuracy and precision
由表7 可以看出,各食品基质的平均回收率在99.97%~109.83%之间,相对标准偏差0.41%~2.65%,均符合要求[16]。验证结果表明该方法具有良好的准确度和精密度。
2.5 代表性样品基质的测定
为了进一步验证该方法的基质适用性以及考察不同食品基质中氟含量水平,选择不同类别的植物性食品基质分别进行氟含量的测定,结果见表8。
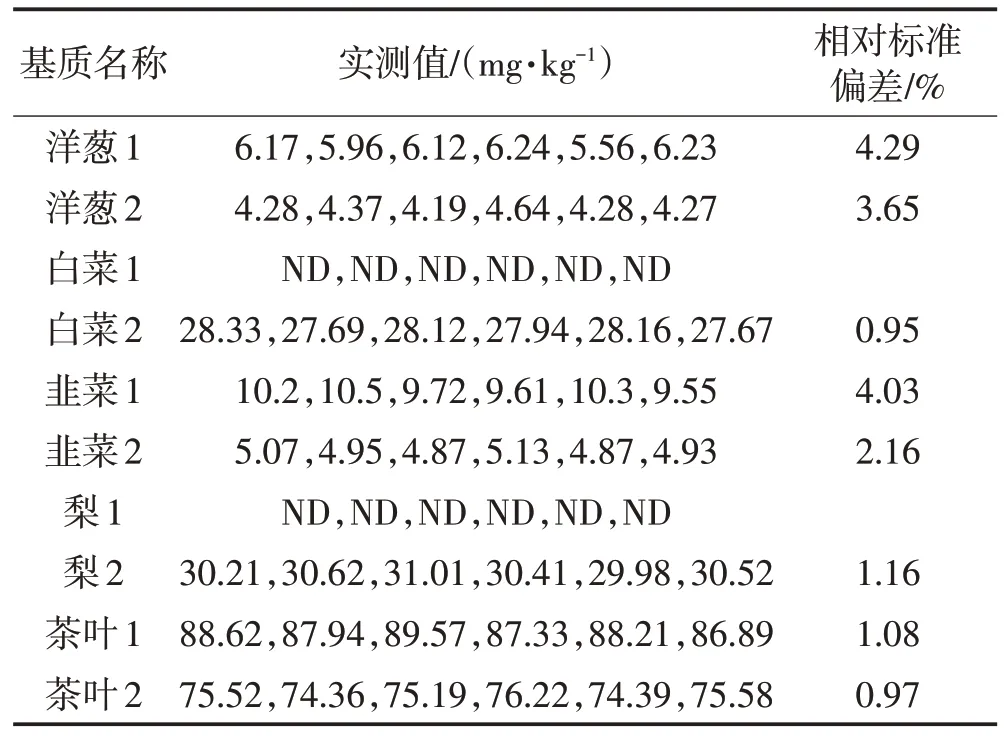
表8 代表性基质测定结果Table 8 Representative matrix determination results
由表8可以看出,采用本研究优化的方法对不同植物性食品基质中的氟含量进行测定,适用性和精密度良好。从不同植物性食品基质的测定结果可以看出,氟化物在植物性食品中的含量总体水平较低,氟暴露的风险较低。
3 讨论与结论
本方法根据氟离子选择电极法的测定原理,通过对GB/T 5009.18—2003[10]中线性范围的验证,推翻了原标准中规定的标准曲线适用范围,并通过进一步优化验证得到了符合理论要求的标准曲线线性范围为0.20~10.00 mg/L,从而避免了由于定量不准确给测定结果带来的误差。
由于GB/T 5009.18—2003[10]中规定样品适宜的称样量为1 g,称样量较小,称量范围较窄,容易给测定的准确度带来影响,并且该标准中没有规定方法的定量限,在实际检测过程中给检验带来了不便,并且称样量过小使得方法的定量限较高。因此,本研究从称样量、称样范围方面进行了优化验证,最终得出结论,对于除茶叶外基质称样量范围为2.5~5.0 g,茶叶基质适宜称样量范围为0.2~0.5 g。通过验证得出,该方法对于除茶叶外的基质,当称样量为2.5 g,定容体积为50 mL 时,方法定量限为4.00 mg/kg;茶叶基质称样量0.2 g,定容体积为50 mL 时,方法定量限为50.00 mg/kg。
本研究在不同植物性食品基质中进行了验证,证明该方法适用于植物性食品基质。通过对代表性基质的测定发现,植物性食品基质中氟化物总体含量水平不高,氟暴露风险相对较低。
除了植物性食品基质外,人们日常饮食对动物性食品的需求量也日益增长,对于动物性食品基质中氟含量的测定方法及含量水平有待进一步的优化及验证。我国作为一个地方性氟中毒病比较严重的国家,加强对食品中氟的监控、降低氟暴露的风险仍十分必要,本研究的开展也在一定程度上对氟中毒疾病的预防起到了促进作用。
