CO2 电还原生成甲酸盐机理及催化剂研究
2023-11-21赵萍萍廉红蕾
赵萍萍,廉红蕾,*
(1.郑州大学化工学院 先进功能材料制造教育部工程研究中心,河南 郑州 450000;2.炼焦煤资源绿色开发全国重点实验室,河南 郑州 450000)
全球变暖是人类社会当前正面临的一个严重问题,这在一定程度上限制了人类社会的发展。CO2作为全球变暖的主要因素,在近年来受到越来越广泛的关注。据2021 年全球碳排放量监测结果显示,2021 年全球CO2排放量增长至34.9 Gt(1 Gt=109t),相比于2020 年增长了4.8%[1],并且呈现出不断增长的趋势。控制CO2排放的途径主要有两种:一是将CO2捕集并封存于地壳,但这种途径较为昂贵费力;二是将CO2作为反应物重新进入碳循环,生成有价值的化学品[2]。其中,第二种途径既能减少其排放,还能生成低碳化学品和燃料[3-5],因此,受到研究者们的青睐。
目前,CO2催化转化技术主要分为四种:生物转化、光电催化、热催化加氢和电化学还原[6]。生物转化是通过模拟自然界的光合作用实现碳循环,但低转化效率限制了该技术的实际应用[7];光电催化过程中,有限的用于氧化还原反应的电子-空穴数量是限制光电催化效率的主要因素[8];热催化加氢是通过加氢过程将CO2转化为CH4、CH3OH 等物质,但高温高压的反应条件又加剧了碳排放[9]。相比之下,电催化CO2还原具有反应条件温和、体系结构简单、能够很好地与其他可再生能源(如太阳能、风能等)耦合使用等优点[10,11],因此,成为CO2减排最清洁和最有效的途径之一。然而,电催化CO2技术仍存在许多限制。第一,CO2的热力学性质非常稳定,其活化需要克服较高的反应势垒,而电催化CO2还原(CO2electrochemical reduction,CO2-ECR)的第一步就是活化CO2分子,因此该过程十分重要;第二,常温常压下,CO2在水系电解液中的溶解度非常低,催化剂表面对CO2分子的吸附十分困难,这很大程度上影响了CO2的传质;第三,CO2电还原过程涉及多个质子和电子转移步骤,生成的产物较为复杂,且水系电解液中还存在竞争性析氢反应(Hydrogen evolution reaction,HER),尤其在较高的过电位下表现得更为明显,这严重限制了其产物选择性。因此,亟需设计并筛选廉价且高效的电催化剂用于电催化CO2还原,增强活性位点对CO2分子的有利吸附,提高还原产物的选择性,并增强催化剂长期使用过程中的稳定性。
CO2电催化还原产物可根据产物含C 数目分为C1 产物和C2+产物,目前,关于C1 产物(CO,HCOOH)的研究相对成熟,在低的过电位下可得到很好的效果,但反应电流密度难以达到预期效果,而C1 产物(CH3OH,CH4)形成过程相对复杂,反应过程法拉第效率(Faradaic efficiency,FE)较低,C2+产物也存在法拉第效率低,反应机理不明确等问题。甲酸作为CO2电化学还原的C1 产物,具有环境友好、无毒、便于分离和储存的优点,可用于多种化工产品的制造[12]。同时,相对于其他CO2-ECR 产物的多电子和多质子转移(如甲烷涉及八个电子和八个质子转移,乙醇涉及12 个电子和12 个质子转移),CO2电催化生成甲酸盐过程只涉及两个电子和两个质子转移,热力学上更容易实现,反应所需活化电位相对较低[13],且在较低的过电位下即可实现很好的法拉第效率。本工作介绍了CO2电还原生成甲酸盐的催化剂类型、反应机理,以及从所使用的反应器类型角度来描述其研究进展。
1 CO2 电还原生成甲酸盐反应机理
电还原CO2生成甲酸盐过程主要涉及三种反应中间体,分别为羧基中间体(COOH*)、甲酸酯中间体(OCHO*或HCOO*)和活性氢物种(H*)。电化学还原CO2生成甲酸盐的多步骤机理如图1所示[14],甲酸盐的生成是通过质子/电子对转移来进行的。首先形成COOH*或OCHO*,接着COOH*或OCHO*通过后续的质子/电子对转移被还原为HCOOH。大量研究表明,OCHO*是甲酸盐生成的关键中间体[15-18],而COOH*中间物种更有利于CO 的生成[19-21],因此,该反应过程存在CO2生成CO 的竞争反应。在反应过程中,可以通过对催化剂进行设计来稳定甲酸盐路径中间体(OCHO*),优化OCHO*在催化剂表面的吸附和解吸强度,从而实现CO2高效转化为甲酸盐。

图1 CO2-ECR 生成甲酸盐、CO 和其他产物的可能反应路径[14]Figure 1 Possible reaction pathways for CO2-ECR to formate,CO and other products[14](with permission from Wiley publication)
目前,用于电催化CO2还原生成甲酸盐的催化剂有多种类型,但实现其可营利的工业化仍具有挑战性。经济技术分析表明,要实现可营利的工业化,甲酸盐电流密度至少为500 mA/cm2,同时其法拉第效率达到70%以上[22]。通过研究CO2电催化转化为甲酸盐的反应机理,探明实际活性位点,从而设计开发高效电催化剂可以为工业级甲酸盐生产奠定坚实基础。
Deng 等[23]通过密度泛函理论(Density functional theory,DFT)研究了金属Bi 和Bi2O3上的CO2电还原反应机理,并分析反应路径,如图2 所示。研究发现,Bi-O 结构的存在降低了CO2的吸附能和中间产物自由能,相比于金属Bi,吸附态CO2更容易吸附在Bi2O3或Bi2O3衍生物表面上。吸附的CO2获得初始电子,形成吸附的 C中间体,C中间体氢化形成OHCO*中间体,接着OHCO*接收电子和质子形成吸附态的HCOOH,并从催化剂表面解吸。金属Bi 与Bi2O3的区别之处在于,金属Bi 的速率决定步骤由初始单电子转移步骤控制,而Bi2O3和Bi2O3衍生物的速率决定步骤是 C中间体的氢化过程。
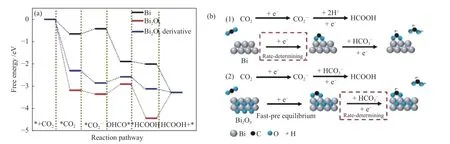
图2 (a)Bi、Bi2O3 和Bi2O3 衍生物上CO2 电还原为甲酸自由能;(b)Bi 和Bi2O3 上CO2 还原过程示意图[23]Figure 2 (a) The free energy calculation of CO2 conversion to formate over Bi,Bi2O3,and Bi2O3 derivative;(b) reduction process of CO2 over Bi and Bi2O3[23](with permission from ACS publication)
DFT 在探明催化反应活性位点方面的应用已较为广泛,但是在高的过电位下,催化剂会产生严重的电化学腐蚀从而发生原位重构,导致实际过程的活性位点可能与DFT 预测的不符。随着原位技术的发展,研究人员不再满足于只借助DFT 来计算得到活性位点,逐渐将原位表征技术和电化学仪器耦合,以实时观察催化剂的表面结构和表面反应[24-26],从而了解催化机理以及催化剂的结构和电还原性能之间的构-效关系。
Li 等[27]利用原位衰减全反射红外吸收光谱实时监测不同电位下六元高熵合金(PdCuAuAgBiIn HEAAs)和六元高熵纳米粒子(PdCuAuAgBiIn HEAPs)中CO2-ECR 反应中间体的演化,以确定CO2高效转化为甲酸盐的机制,如图3 所示。原位红外光谱表明,当施加电位从开路电压(Open circuit potential,OCP)增加到-0.3 V vs.RHE 时,在1615、1534、1404、1368、1270、1173 cm-1处开始出现几个新的谱带,其中,1534、1404、1067 cm-1处的谱带与催化剂表面的多齿吸附甲酸盐(bi-HCOO*)相关,随着施加电位的增加((-0.3)-(-0.5)V vs.RHE),Bi-HCOO*谱带明显增强;1615、1368、1171 cm-1处的谱带归属于单齿吸附甲酸盐(m-HCOO*),随着施加电位的继续增大((-0.3)-(-1.1) V vs.RHE),m-HCOO*谱带持续增强。通过对原位红外谱图分析,得到CO2电还原生成甲酸盐的反应机理:首先,CO2吸附在PdCuAuAgBiIn HEAAs 表面形成*CO2物种,*CO2进一步加氢生成多齿吸附甲酸盐(bi-HCOO*),然后bi-HCOO*随着施加电位的增强发生构型转变形成单齿吸附甲酸盐(m-HCOO*),并从催化剂表面解吸得到甲酸盐。而在PdCuAuAgBiIn HEAPs 表面,部分m-HCOO*会加氢脱水形成*CHO,并进一步氢化为CH4,导致甲酸盐选择性下降。

图3 (a)PdCuAuAgBiIn HEAAs 和(b)PdCuAuAgBiIn HEAPs 的原位衰减全反射红外吸收光谱谱图[27]Figure 3 In-situ attenuated total reflectance infrared absorption spectroscopy of (a) PdCuAuAgBiIn HEAAs and(b) PdCuAuAgBiIn HEAPs[27](with permission from Wiley publication)
Duan 等[28]利用原位电化学拉曼光谱对Bi基催化剂(BiCuSeO)上的CO2-ECR 过程和中间产物进行了研究,原位拉曼装置如图4(a)所示,重点监测了不同还原电位和还原时间下的CO2-ECR 过程(图4(b)–(d))。研究发现,在-0.5 V vs.RHE 或更低电位下,在1160 和1540 cm-1处检测到两个明显的拉曼峰,其中,1160 cm-1处的峰归属于CO2-ECR 过程中表面吸附碳酸盐的C=O 伸缩振动,1540 cm-1处的峰归属于OCHO*的不对称C-O 伸缩振动,这两个峰都归属于CO2-ECR 过程中甲酸盐形成的关键中间体OCHO*,证实了CO2可以在较低的电位下直接被活化并转化为甲酸盐。这两个峰的峰强度随着施加电势的增加而逐渐升高,在-0.7 V vs.RHE 时达到最高,该变化趋势可能与中间产物的吸附与转化有关。此外,由于甲酸盐中间体良好的吸附和质子俘获能力,其峰强度随着反应的进行而逐渐增强。没有检测到与*COOH 中间体的C–O 拉伸或C=O 拉伸相关的谱带,表明在BiCuSeO 上几乎没有CO 形成。
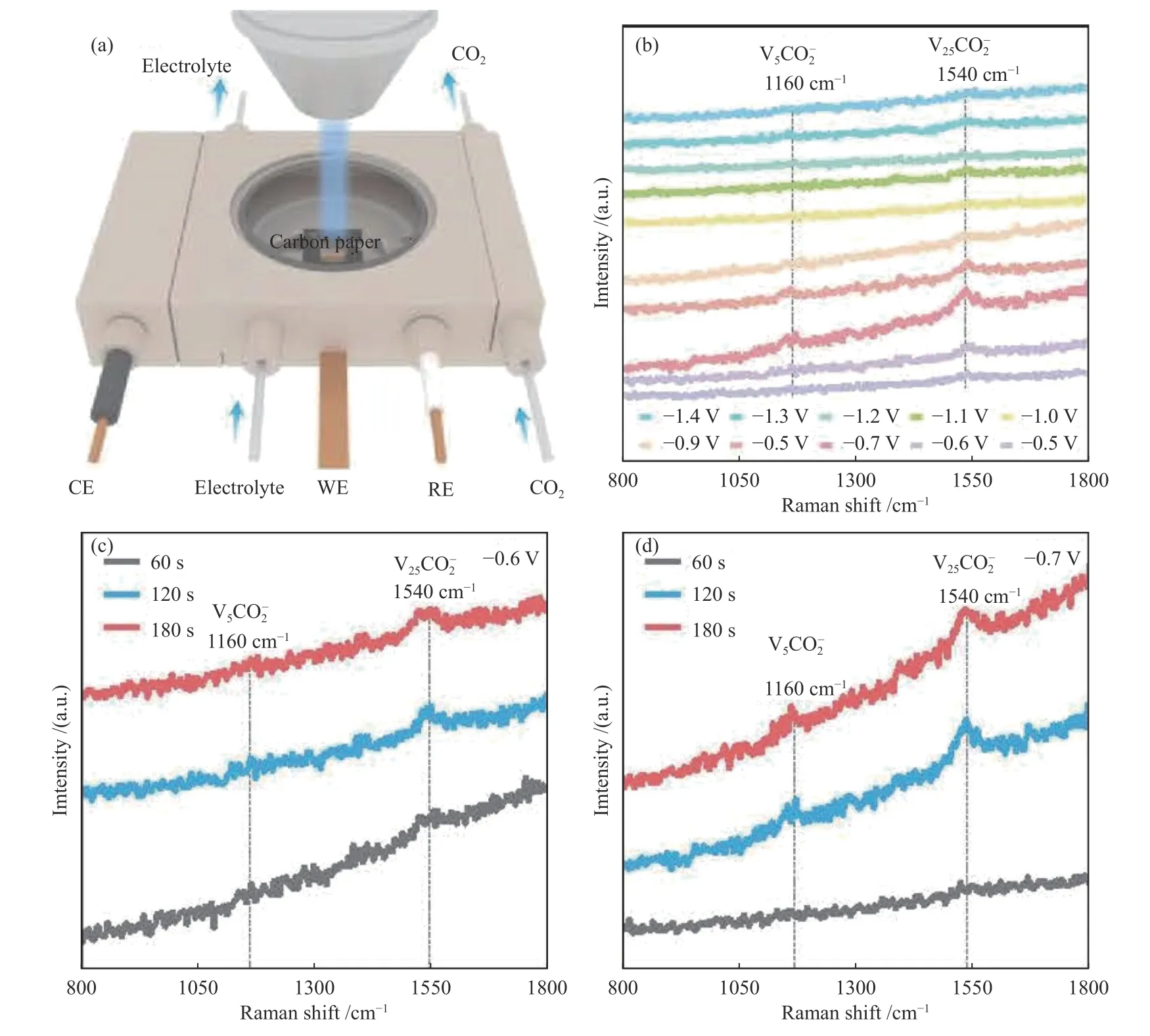
图4 (a)原位拉曼测试装置示意图;在0.5 mol/L KHCO3 电解液中,CO2 鼓泡条件下,BiCuSeO 催化剂的原位拉曼光谱谱图:(b)不同还原电位,((c)-(d))不同反应时间[28]Figure 4 (a) Schematic illustration of the in situ Raman measurement device during the CO2-ECR;(b) potential-dependent and((c)-(d)) time-dependent in situ Raman spectra of BiCuSeO catalysts in 0.5 mol/L KHCO3 solution under CO2 bubbling[28](with permission from Nature publication)
目前,已有越来越多的研究将原位表征技术与电化学仪器耦合来实时监测反应中间体,如借助原位XRD、原位红外(薄层模式和衰减全反射模式)、原位Raman、原位同步辐射等表征技术明确催化反应机理,同时辅以DFT 密度泛函理论计算,完善理论模型,进一步探究特定活性位点对催化剂性能的影响作用,这对高效催化剂的设计合成具有深远意义。
2 催化剂类型
在对CO2电还原制备甲酸盐反应机理的研究中,人们提出该过程主要包含两种机制。一种是活性位点与CO2的C 结合,生成羧基中间体(COOH*),更倾向于形成CO;另一种是活性位点与CO2的O 结合,生成甲酸酯中间体(OCHO*),更倾向于生成HCOOH[29,30]。活性位点与CO2的C 结合还是与CO2的O 结合,主要取决于催化剂的性质。一般来说,Bi、Sn、In、Pb、Hg、Tl、Cd 等金属更倾向于与O 结合生成HCOOH,而Au、Ag、Zn、Ni 等金属更倾向于与C 结合生成CO[31,32]。但基于环境友好和催化剂储量及成本问题的考量,目前,研究最为广泛的还是一些低成本且无毒的过渡金属基催化剂,如金属Bi 催化剂、Sn 催化剂和In 催化剂等。根据催化剂组成不同,作者将催化剂类型划分为金属催化剂、原子分散催化剂、金属氧化物、炭材料和复合材料催化剂。
2.1 金属催化剂
金属催化剂是指金属作为主要活性组分在反应过程中发挥催化活性的一类催化剂。金属催化剂按照其组成成分的不同,可分为单金属催化剂和合金催化剂。单金属催化剂即只含有一种金属组分的催化剂,其结构较为简单,且具有优异的导电性能,目前,已被广泛的应用于电催化CO2还原过程。
Luo 等[33]课题组以Cu 网作为衬底进行电沉积得到三维分级多孔In 催化剂(hp-In),该结构有利于提供有效的气体传输通道,以最大程度的吸附CO2。电化学研究发现,在整个测试电位范围内,hp-In 催化剂上H2的法拉第效率都被抑制在10%以下,这归因于阴极反应导致的质子消耗提高了电极和电解质界面处的局部pH,降低了HER 活性;同时,hp-In 催化剂上电还原的总电流密度也明显高于In 箔,随着电位增加,hp-In 催化剂在含有0.1 mol/L KHCO3电解液的H 型电解池中,-1.2 V vs.RHE 时,电流密度可达67.5 mA/cm2,这主要是由于其多孔结构提供了较大的电化学活性表面积。Greeley 等[34]对电催化材料的筛选发现,金属Bi 催化剂可以在抑制竞争性HER 的同时生成甲酸盐,根据DFT 密度泛函理论计算,Bi 表面的氢气吸附自由能较高,从而抑制产氢。同时,金属Bi表面对 C中间体的亲和力适中,因此,C可以弱吸附在Bi 表面,与H2O 快速反应生成HCOO-[35,36]。Qiao 等[37]开发出超薄Bi 纳米片(Bi NS),并在强酸介质中电还原CO2,采用0.05 mol/L H2SO4作电解质溶液以抑制碳酸盐的形成,同时通过加入3 mol/L KCl 引入K+离子调控局部环境。在流动池中,施加-1.28 V vs.RHE 电位时,甲酸生成的法拉第效率为92.2%,同时达到237.1 mA/cm2的高电流密度,高于200 mA/cm2的工业化电流密度[38]。值得注意的是,在强酸性介质中添加KCl 后,HER明显被抑制,CO2电还原为甲酸盐的能力显著增强。这是因为K+降低了质子覆盖率进而抑制竞争性HER,降低酸中CO2还原的能垒限制从而稳定*OCOH 反应中间体(如图5 所示),同时超薄的Bi 纳米片增强了CO2的传质,有助于实现高效电还原CO2生成甲酸。同时,甲酸在酸性溶液中直接生成,代替从甲酸盐中再生,从而简化下游分离设备,显著节约成本。

图5 (a)基于Poisson-Nernst-Planck 模拟得到的H + 离子浓度CH+ 与外亥姆霍兹面之间距离的关系;(b)不同K + 浓度下的产物选择性;(c)在没有或存在K + 的情况下,CO2-ECR 生成HCOOH 的吉布斯自由能[37]Figure 5 (a) The ions concentration CH+ as function of distance from the outer Helmholtz plane (OHP) based Poisson-Nernst-Planck simulations;(b) product selectivity at different K + concentrations;(c) Gibbs free energy diagram of CO2-ECR to HCOOH in the absence or presence of K + cations[37](with permission from ACS publication)
相对于单金属催化剂有限的催化能力,双金属合金催化剂电还原CO2性能大为提升,这是因为金属杂原子的加入可以调控金属d带空穴,不同金属间的电负性差异还导致金属间更容易发生电荷转移,加快电极表面电子传输,改善反应动力学,同时调控催化剂表面对中间体物种的亲和力,使CO2电还原更倾向于甲酸路径[39]。
Li 等[40]对比了纯Cu 催化剂和CuSn 合金催化剂的电化学性能。研究发现,CuSn 合金吸附CO2的能力显著强于纯Cu,且在纯Cu 中引入最少量的Sn 时,CO2-ECR 性能就显著提高。结合原位拉曼光谱发现,CuSn 合金在1355 cm-1处出现HCOO*的峰,而纯Cu 的原位拉曼光谱没有HCOO*信号,表明双金属间的协同作用促进了HCOO*关键中间体的形成。DFT 密度泛函理论计算表明,形成合金后,Sn 位点处甲酸盐形成的能垒显著降低,而单金属催化剂上由于H2形成的能垒较低,因此HER 占主导。Ren 等[41]采用电沉积法制得了表面褶皱诱导的Sn-Bi 双金属界面结构,其形貌及性能如图6 所示。在含0.5 mol/L KHCO3电解液的H 型电解池中,-0.84 V vs.RHE 时,FEHCOO-为(96.4±2.5)%。优异的电催化性能归因于Sn-Bi 界面结构中,Sn倾向于将电子从p轨道转移给界面Bi 原子,相比于纯Sn 催化剂,Sn-Bi 界面结构具有适宜的价电子,导致减弱的Sn-C 杂化(竞争COOH*路径)和适宜的Sn-O 杂化(HCOO*路径),从而可以最好地平衡两种竞争反应中间体(HCOO*和COOH*)的吸附,并且表现出最佳的甲酸盐选择性,再次验证了双金属催化剂相对于单金属催化剂具有更优异的催化性能。

图6 Sn-Bi 界面结构催化剂的(a)扫描电镜照片;(b)透射电镜照片;不同样品(Sn-Bi界面,Sn-Bi 合金,电沉积ED-Bi,水热法SnOx,电沉积ED-Sn)的(c)FEHCOOH 和(d)PCDHCOOH[41]Figure 6 (a) SEM image and (b) TEM image of Sn-Bi interfacial catalyst;(c) FEHCOOH and (d) PCDHCOOH over various samples (Sn-Bi interface,Sn-Bi alloy,electrodeposition ED-Bi,hydrothermal SnOx and electrodeposition ED-Sn)[41](with permission from Nature publication)
近年来,高熵合金以其优异的物理、化学和力学性能,成为金属合金领域的研究热点之一。相比于传统合金的某一种组分占主导,高熵合金中没有任何一种元素占据主导地位,各元素的比例较为平均,一般为5%-35%,通常组元数≥5[42]。高熵合金因其高温稳定性及抗氧化性能在CO2-ECR 领域具有很大的应用潜力。虽然合金化是提高CO2-ECR 目标产物选择性的有效策略,但对于高熵合金来说,仍伴随着制备过程较为复杂、能耗大、制备的合金材料尺寸大等缺点。同时,研究发现,等摩尔组合物通常最容易获得,但其往往并不会具有最好的性能[43],因此,调控高熵合金的微观成分和设计开发新型的CO2-ECR 电催化剂至关重要。
Li 等[27]采用冻融法合成PdCuAuAgBiIn 高熵合金气凝胶(HEAAs),研究发现,该催化剂同时兼具了高熵合金的高机械强度和气凝胶的高孔隙率等优点,多孔结构中观察到大量的缺陷,这可以提供更多的不饱和位点,有利于CO2分子的活化。电化学行为研究发现,PdCuAuAgBiIn HEAAs在较宽的电位窗口内对产物HCOOH 和C1 产物的选择性生成表现出优异的性能。在含0.1 mol/L KHCO3电解液的H 型电解池中,(-0.7)-(-1.1) V vs.RHE 电位时,C1 产物的FE 接近100%;在-1.1 V vs.RHE 电位下,HCOOH 的FE 达到最大值(98.1%)。此外,在含0.5 mol/L KHCO3电解液的流动池中可实现接近200 mA/cm2的电流密度,优异的电化学行为归因于PdCuAuAgBiIn HEAAs 中Pd 金属的不饱和配位。扩展X 射线吸收精细结构表征结果显示,PdCuAuAgBiIn HEAAs 中Pd 配位数(为9.9±1.1)相对于Pd 箔(配位数为12)有明显下降。这种不饱和配位通过调节不同金属的电子结构来调控催化剂表面对不同中间体的亲和力,增强对OCHO*中间体的吸附,并抑制CO 中毒和竞争性HER,如图7 所示。
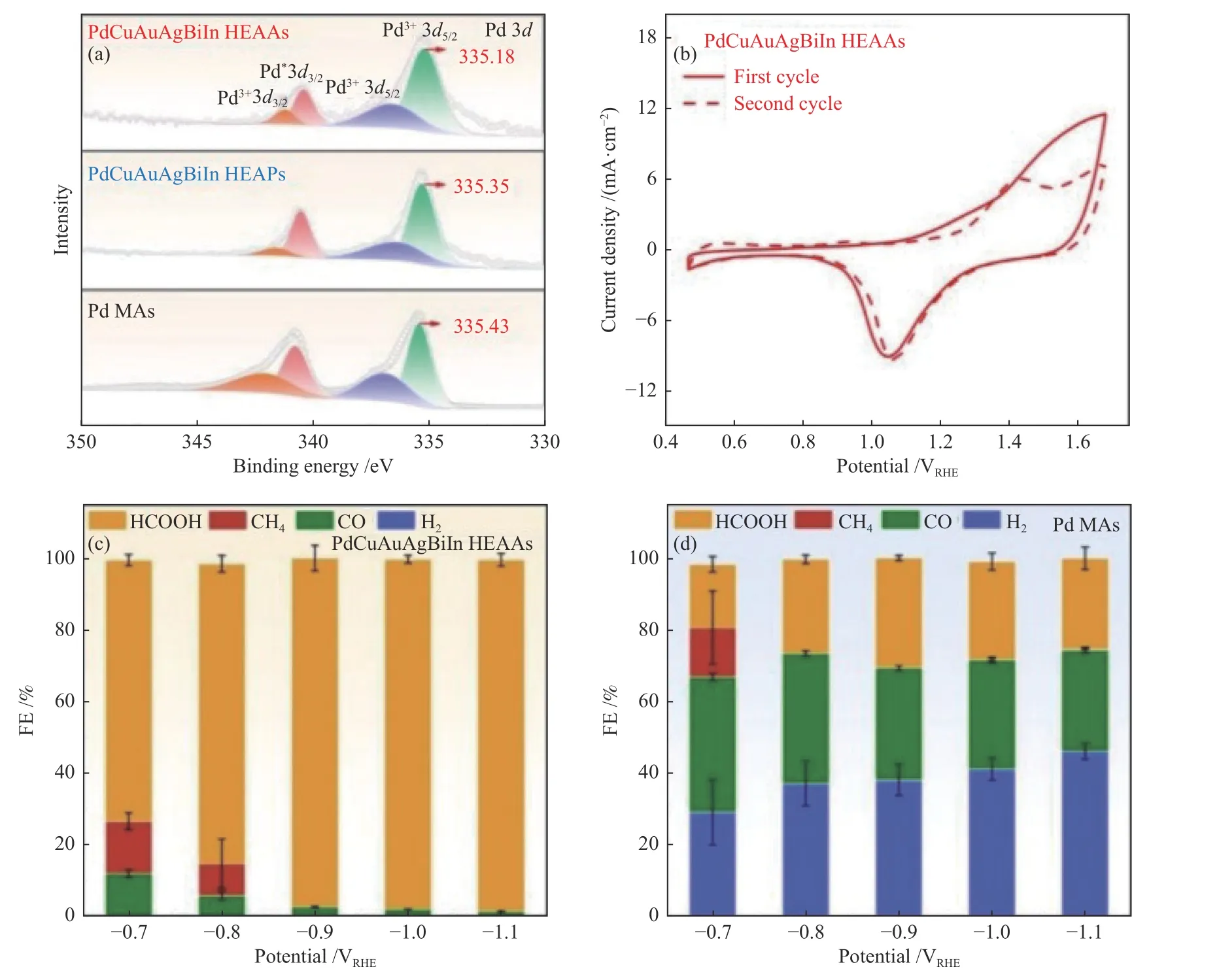
图7 (a)Pd 3d 精细XPS 光谱谱图;(b)PdCuAuAgBiIn HEAAs 的伏安曲线;(c)PdCuAuAgBiIn HEAAs 和(d)Pd 金属气凝胶(Pd MAs)在不同电位下各种产物的法拉第效率[27]Figure 7 (a) Pd 3d fine XPS spectra;(b) CO stripping curve of PdCuAuAgBiIn HEAAs;Reduction potential dependent FEs measured on (c) PdCuAuAgBiIn HEAAs and (d) Pd MAs[27](with permission from Wiley publication)
2.2 原子分散催化剂
通常来说,金属颗粒的尺寸是影响其催化活性和原子利用率的关键要素,减小颗粒粒径有利于调节金属的电子结构和暴露更多的活性位点,催化活性和原子利用率也越高[44]。当粒径小到金属以原子形式分散时,此时的催化剂具有最高的原子利用率和更均匀的活性位点,并且表现出非凡的催化性能[45-47]。原子分散催化剂根据中心原子的数量,可以分为单原子催化剂、双原子催化剂和原子簇催化剂[48]。单原子催化剂是指由单个金属位点作为活性中心的催化剂,单个金属位点往往与载体的其他原子进行配位从而调控其电子结构[49]。
Zhang 等[50]以ZnO 纳米片为载体分散金属Sn,得到Sn 掺杂量仅为1.2%的Sn SA/ZnO 催化剂。该催化剂呈现2D 多孔纳米片结构,平均厚度只有21 nm,有利于暴露更多的活性位点和增强CO2-ECR 传质,单原子和金属氧化物载体之间的强相互作用可以调节Sn 单原子的电子结构,同时还能抑制其团聚,从而明显提高本征活性和电化学稳定性。在含有0.5 mol/L KHCO3电解液的流动池中进行电化学测试,发现在(-0.7)-(-1.7) V vs.RHE电位时,Sn SA/ZnO 上甲酸盐的FE 始终大于单组分的ZnO 和Sn。Sn SA/ZnO 中的ZnO 载体可以更好地暴露活性位点从而增强活性组分对CO2分子的有效捕获和活化,在Sn SA/ZnO 电催化剂表面形成稳定的OCHO*中间体也是其高性能的主要原因。
Lu 等[51]以金属In 作为活性中心,分散在氮掺杂碳骨架上,得到In-N-C 电催化剂,如图8 所示。由于金属和碳基载体之间高速的电子转移和高度暴露的In 活性位点,In-N-C 对CO2还原为甲酸盐具有较高的催化活性。在含有0.5 mol/L KHCO3电解液的H 型电解池中进行电化学研究,发现在-0.59 V vs.RHE 电位时就有甲酸盐生成,并在-0.79 V vs.RHE 时FEHCOO-达到最大值(80%),其电流密度为6.8 mA/cm2。

图8 (a)*OCHO 和*COOH 在In-N-C 表面的稳定吸附的几何结构;(b)在In-N-C、In(101)、In(110)和In(112)表面CO2-ECR 自由能[51]Figure 8 (a) Stable adsorption geometry of *OCHO and *COOH on the surface of In-N-C;(b) free energy diagram on the surface of In-N-C,In(101),In(110) and In(112) under CO2-ECR[51](with permission from ACS publication)
相比于单原子催化剂,在双原子催化剂中,引入与初级金属中心相邻的次级金属中心,两者共同承担活性中心的作用,这改善了单个活性中心无法提供多位点吸附的限制,且次级金属中心还可以调节初级金属中心的电子结构,更有利于催化反应过程[52-56]。为了进一步地改善吸附位点的限制,向双原子催化剂中引入一个或者多个原子以形成原子簇催化剂,原子簇材料包含了更多的金属原子,相对于单原子和双原子,其具有更高的金属负载量,并能够提供更多的活性中心。此外,原子簇特有的金属中心配位环境通常会产生独特的协同效应,使其更有利于CO2电还原过程[57]。
Jiang 等[58]合成了碳负载的氧化铋簇催化剂用于电催化CO2还原反应。DFT 结果显示,在氧化铋簇上,OCHO*中间体形成的自由能明显低于COOH*和H*,说明氧化铋簇更有利于OCHO*关键中间体的稳定,同时抑制其他含碳产物的生成,在膜电极池中,电解液为0.1 mol/L KOH 的条件下进行电化学测试,在-1.2 V vs.RHE 电位下,CO 的FE 仅为3.4%。电化学行为研究发现,当电流密度高达500 mA/cm2时,甲酸盐的FE 保持在90%以上,对应于450 mA/cm2的甲酸盐电流密度,高于实现工业化要求的电流密度[38]。
2.3 金属氧化物催化剂
金属氧化物催化剂中的金属-氧(M-O)结构可以稳定电还原过程中的关键中间体,还能促进反应动力学,氧空位的存在也有利于催化剂表面结合CO2的O 而表现出更倾向于甲酸盐路径,从而使金属氧化物催化剂在电催化领域显示出较高的催化性能。
Miao 等[59]合成了纳米颗粒状的Bi2O3(Bi2O3-A)和纳米棒状的Bi2O3(Bi2O3-B),用于电催化CO2还原为甲酸盐,并探究催化剂涂覆量对CO2还原的影响。结合Tafel 动力学测试结果,发现相比于Bi2O3-B,Bi2O3-A 的Tafel 斜率更小,为166 mV/dec,表明Bi2O3-A 的反应动力学更快,具有更优异的电催化性能。在H 型电解池中,电解液为0.5 mol/L KHCO3条件下,在-1.2 V vs.RHE 电位下生成甲酸盐的最大部分电流密度为22 mA/cm2,是Bi2O3-B(8.9 mA/cm2)的三倍,甲酸生成的FE 为91%,且在长达23 h 的电解过程中FE 和部分电流密度几乎保持稳定。作者通过增大催化剂的涂覆量,探究活性组分含量对CO2-ECR 的促进关系。涂覆量越大,活性组分的含量越高,这从产物FE 和部分电流密度的增加就能明显看出,但过大的涂覆量导致催化剂层过厚,在电解过程中容易脱落,这导致了催化性能的快速下降。
Chen 等[60]采用水热反应法制备了如图9 所示的波状SnO2材料(NW-SnO2),并用商业SnO2纳米粒子(NP-SnO2)作对比,通过X 射线光电子能谱(XPS)对比两种不同样品发现,NW-SnO2和NPSnO2光谱中都显示晶格氧和氧空位共存,但NWSnO2的氧空位含量要明显高于NP-SnO2,显示出较大的差异。同时,XPS 还证实了O–Sn–O 键的存在,不同的是,NW-SnO2的Sn 3d5/2结合能要低于NP-SnO2,表明电荷从氧空位转移到Sn,更有利于催化反应的进行。电化学测试结果显示,在含有0.5 mol/L KHCO3电解液的H 型电解池中,在-1.0 V vs.RHE时,C1 产物的总FE 接近96%,H2的FE 被抑制到4%以下,CO2电还原为HCOOH 的局部电流密度可达22 mA/cm2,在-1.0 V vs.RHE 时,FEHCOOH最大值为87.4%,高于NP-SnO2催化剂的性能。

图9 NW-SnO2 的(a)低分辨率和((b)、(c))高分辨率透射电镜照片[60]Figure 9 (a) Low resolution TEM images and ((b),(c)) high resolution TEM images of NW-SnO2[60](with permission from Elsevier publication)
2.4 炭材料
碳基材料因其储量丰富、比表面积大、成本低、具有可调节的电导率和优异的电化学活性[61]等优点也被广泛应用于CO2-ECR,且相比于金属材料,碳基催化剂更耐中毒[62],且可组装为多种尺寸和结构,如碳纳米球、碳纳米纤维、碳纳米管、洋葱碳、石墨烯、金刚石等[63]。在碳基材料中引入杂原子(如N、P、S、B 等)[64-66],可以调节电子结构,增加活性位点数量,降低CO2-ECR 关键中间体的自由能。Natsui 等[67]开发了B 掺杂金刚石(BDD),在含有0.5 mol/L KCl 电解液的流动池中甲酸盐的法拉第效率超过94.7%。优异的性能归因于B 原子对金刚石的掺杂,它们对OCHO*中间体具有选择性,同时还能抑制HER。此外,还可以通过杂原子共掺杂策略,如N、S 共掺杂[68]、N、P 共掺杂[69]、B、N 共掺杂[70]等引入缺陷和调节电子结构,进一步提高催化活性。
除了杂原子掺杂炭材料用作CO2电还原的催化剂之外,还衍生出炭材料负载的金属催化剂,其在提高产物选择性、降低还原过电位等方面也表现出优异的性能[71]。Yang 等[72]合成了一种金属Bi 掺杂的水热碳改性碳纳米管催化剂(Bi-HCT/OCNTs)。该催化剂在含有0.5 mol/L KHCO3的H 型电解池中显示出优良的催化性能,在-1.01 V vs.RHE 时,甲酸盐的选择性为94.8%。催化剂表面的Bi-C 位点有利于捕获CO2分子,促进OCHO*关键中间体的生成,从而调节产物分布。Bi-HCT/OCNTs 中的水热碳以碳层的形式均匀包裹碳纳米管,催化剂具有丰富的互连通道,这可以加快反应的传质,同时,炭材料良好的导电性也有利于电子的转移,这些原因共同导致了Bi-HCT/OCNTs的高催化性能。
Ning 等[73]在还原氧化石墨烯(RGO)上原位制备超薄硫化铟纳米片,得到In2S3-RGO 催化剂。由于RGO 的存在,In2S3的厚度从30.2 nm 减小到3.9 nm,这在很大程度上促进了活性位点的暴露。RGO和In2S3组分间发生协同作用,C=O 基团与In3+耦合形成C–O–In 键,降低了In 表面的自由能,有利于结构的稳定。DFT 理论计算发现,完全暴露的In2S3(440)面对催化甲酸盐的生成表现出更高的活性。在含有0.1 mol/L KHCO3的H 型电解池中,对该催化剂的电化学性能进行测试,发现在-1.2 V vs.RHE 电位下,FEHCOO-可高达91%,优异的选择性归因于较大的电化学活性表面积和暴露的高密度的活性位点。但In2S3不稳定,在电解条件下容易发生硫的还原。
Liu 等[74]在疏水性碳纳米纤维(C/HB)和亲水性碳纳米纤维(C/HL)上原位生长氧化铋纳米片,分别得到Bi2O3@C/HB 和Bi2O3@C/HL 催化剂,并在含0.5 mol/L KHCO3电解液的H 型电解池中测试了两者的电催化性能。研究发现,优异的性能源于碳纳米纤维上负载的Bi2O3纳米片,在施加较小的电位时,两种催化剂的电催化性能较为接近,但在较大的施加电位下,Bi2O3@C/HB 的性能明显优于Bi2O3@C/HL,如图10((a)–(c))所示。另外,在含1 mol/L KOH 电解液的流动池中评估了两种催化剂的电催化性能,如图10((d)–(f))所示。流动池中CO2-ECR 的起始电位仅为-0.41 V vs.RHE,且在-1.5 V vs RHE 电位下,Bi2O3@C/HB 催化剂上的甲酸盐部分电流密度即可达到285 mA/cm2。其优异的性能归因于疏水载体减轻了催化剂上的液泛问题,抑制了氢键形成,从而实现优异的甲酸盐选择性。
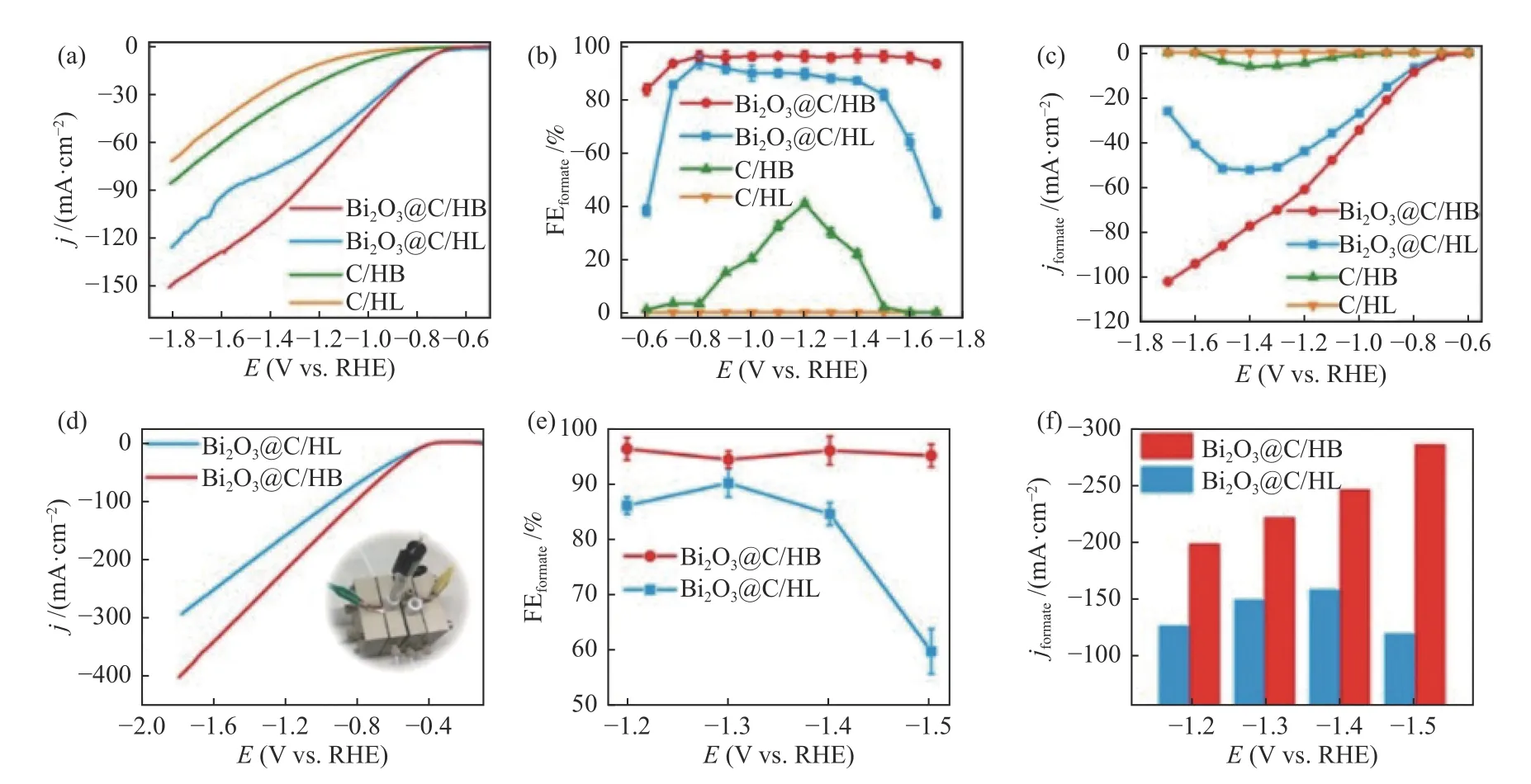
图10 在含0.5 mol/L KHCO3 电解液的H 型电解池中,Bi2O3@C/HB、Bi2O3@C/HL、C/HB、C/HL 的(a)线性扫描伏安曲线,(b)甲酸盐法拉第效率,(c)甲酸盐部分电流密度;在含1 mol/L KOH 电解液的流动池中,Bi2O3@C/HB 和Bi2O3@C/HB 的(d)线性扫描伏安曲线,(e)甲酸盐法拉第效率,(f)甲酸盐部分电流密度[74]Figure 10 (a) Linear sweep voltammetry curve,(b) FEformate,(c) jformate of Bi2O3@C/HB,Bi2O3@C/HL,C/HB,C/HL in a H-cell containing 0.5 mol/L KHCO3 electrolyte,(d) Linear sweep voltammetry curve,(e) FEformate,(f) jformate of Bi2O3@C/HB,Bi2O3@C/HL in a flow cell containing 1 mol/L KOH electrolyte[74](with permission from ACS publication)
金属-有机框架(MOFs)是由有机连接物和金属节点构建而成的一类多孔材料,具有大的比表面积、高的孔隙率和可调节的孔径,能使活性物种分散地更均匀从而提供更多的活性位点[75]。此外,MOFs 材料可用于制备含金属纳米颗粒和多孔炭材料的杂化催化剂,近年来成为电化学还原CO2的优良催化剂前驱体。Deng 等[76]和Wang 等[77]分别通过热解MOFs 前驱体合成碳层限域金属氧化物催化剂,在流动池中(电解液为1 mol/L KOH)甲酸盐电流密度超过100 mA/cm2。优异的性能归因于金属氧化物和碳层的共同作用,氧化物有助于改善反应动力学和提高产物选择性,而碳层有助于提高活性和电流密度,同时碳层限域金属氧化物还有助于降低阻抗,但热解过程会导致MOFs的多孔结构坍塌,导致比表面积下降和不利于调节孔径。
2.5 复合材料
电催化活性在很大程度上取决于催化剂的化学成分,开发具有独特功能化结构和大量催化活性位点的催化剂对提高催化活性具有重要意义。目前,常用的策略是通过引入其他组分,如金属、金属氧化物、炭材料等[78-82],与活性组分产生协同作用,提高催化剂表面的电子亲和力,这有利于电还原关键中间体的稳定,从而优化CO2-ECR 活性[83]。
催化剂组分的稳定性对催化性能的提高具有重要意义,Lin 等[84]以Bi2S3作为催化剂前驱体进行研究,在流动池中(电解液为1 mol/L KOH),在-0.38 V vs.RHE 较低电位下就有甲酸盐生成,FEHCOO-可达66%;当电位增加到-0.52 V vs.RHE时,FEHCOO-增加到96%。进一步增大施加电位到-0.95 V vs.RHE,总电流密度急剧增大到2 A/cm2,其中,甲酸盐部分电流密度高达1.9 A/cm2,高于目前已知的大多数催化剂性能。优异的催化性能归因于Bi2S3在电还原过程中动态演变为更加稳定的Bi0团簇和Bi2O2CO3形成的复合物。在CO2-ECR 期间,Bi0团簇和Bi2O2CO3共存,并对Bi2O2CO3的表面进行修饰从而改变Bi2O2CO3的电荷分布,Bi2O2CO3的表面上带正电的Bi 原子作为活性位点,与CO2产生强库仑相互作用,从而实现CO2的最佳吸附。此外复合材料在后续氢化形成甲酸盐过程需要克服的能垒也更低,更有利于甲酸盐的形成。
Dou 等[85]制备了SnO2掺杂CuS 和S 形成的CuS/SnO2-S 复合材料,并在含有0.5 mol/L KHCO3电解液的H 型电解池中,将其应用于CO2-ECR 选择性转化为甲酸盐。研究结果表明,CuS 的引入加快了电子传输,从而促进CO2-ECR 过程动力学,同时,S 的引入减少了CuS 的还原,在-0.8 V vs.RHE 电位下,FEHCOO-达到84.9%,甲酸盐部分电流密度约为18.8 mA/cm2,优异的性能是各组分综合作用的结果。Jiang 等[86]构建了纳米管状结构的SnCu 双金属氧化物复合材料(SnCu-CNS),其形貌如图11 所示,并在含0.1 mol/L KHCO3电解液的H 型电解池中进行了电化学性能探究,发现SnCu-CNS 的电流密度明显高于单一组分的SnO2和CuOx,且在(-0.4)-(-1.2) V vs.RHE 电位下,HER 的法拉第效率降低到10%以下。在-0.9 V vs.RHE 电位下,SnCu-CNS 上的FEHCOO-达到最大值(95.1%),优异的性能归因于纳米管状结构可以暴露更多的活性位点,提供更大的比表面积,同时保证了CO2的良好吸附,并增强CO2-ECR 的电子转移。此外,SnCu-CNS 可以形成Snδ+和CuO 中心,降低关键中间体的形成能垒,从而获得高的甲酸盐选择性。

图11 SnCu-CNS 的(a)扫描电镜照片;(b)透射电镜照片[86]Figure 11 (a) SEM and (b) TEM images of SnCu-CNS[86](with permission from Wiley publication)
近年来,众多学者致力于从事CO2电催化还原制甲酸盐的研究。总体上,有目的的构建催化剂结构与优化反应器体系,是电催化性能得到大幅提升的主要原因所在。因此,为了更直观地了解并比对不同催化剂的电催化性能,将上述文中涉及的催化剂及其性能参数汇总于表1。目前,尽管金属基催化剂在电还原CO2产甲酸盐领域已取得了突破性的进展,但仍存在现阶段急需攻克的难题,例如在电还原过程中如何实现尽可能低的起始电位(标准电极电势为-0.12 V vs.RHE)[13]、高电流密度(>500 mA/cm2)、高稳定性(运行时间长达上千小时)[23]的问题,这些仍是现阶段乃至未来的研究重点,需要从CO2-ECR 性能的影响因素入手,深入研究催化剂各组分之间、催化剂与电解液之间的协同机制,设计开发高效电极结构,为工业级CO2电化学产甲酸盐奠定坚实基础。

表1 不同催化剂电还原CO2 生成甲酸盐的性能Table 1 Performance of electroreduction CO2 towards formate over various catalysts
3 CO2 电还原性能影响因素
CO2电还原性能主要通过FE、电流密度、过电位、电化学活性比表面积、稳定性等指标进行评估,其影响因素繁多,总体上可分为催化剂性质、电解液、杂质气氛和反应器的影响。
3.1 催化剂性质
为了提高CO2电还原制甲酸盐的活性和选择性,首先需要对催化剂材料进行设计。例如可通过改变催化剂的形貌、尺寸、引入缺陷等使其暴露更多的活性位点;通过杂原子掺杂来调控金属的电子结构,以及通过对金属特定晶面的可控合成,实现CO2分子的适中吸附和产物的弱吸附,从而稳定反应中间体和降低产物生成过程中各活性中间体的形成能垒,这些研究在改善CO2-ECR 性能方面已经取得了实质性进展。
对催化剂形貌和尺寸的调控一直都是广大研究人员设计催化剂时首要考虑的问题,其主要目的是改善催化剂活性比表面积,增大电极材料与电解液的接触面积从而加快传质速率。Meng 等[87]在柔性三维掺氮碳泡沫上溶剂热生长氧化铋超薄纳米片,三维碳泡沫框架为催化剂提供了较大的比表面积,有利于活性位点的暴露和加快传质,在-1.0 V vs.RHE 电位下,FEHCOO-为94.1%。Wang 等[88]利用溶剂热反应合成了多层结构的S 掺杂BiOCOOH前驱体,并在电化学驱动下原位转变为Bi-S 催化剂。研究发现,电化学转化不改变前驱体的形貌,通过改变S 的含量可以显著改善Bi-S 的尺寸。CO2电还原性能与S 的掺杂量呈火山型关系,S 掺杂量为4.6%时催化性能最好,在含0.5 mol/L KHCO3电解液的H 型电解池中,-0.9 V vs.RHE 电位下,FEHCOO-最大为96.7%;在-1.2 V vs.RHE 电位下,部分电流密度达到25.47 mA/cm2,当S 掺杂量进一步增大,催化剂表面对H2O 的吸附能力增强,可用于电还原生成甲酸盐的活性位点数减少,甲酸盐选择性降低。Qiu 等[89]通过改变二卤素氧化卤化铋(BiOX0.5Y0.5,X,Y=Cl、Br、I)中卤素元素的组合来调控玻碳电极表面生长的纳米Bi 尺寸。研究结果如图12 所示,甲酸盐的电流密度和法拉第效率都表现出显著的尺寸效应,在-1.6 V vs.RHE 电位下,通过BiOCl0.5Br0.5得到的纳米Bi(平均尺寸约8.8 nm)电还原生成甲酸盐的部分电流密度为9.7 mA/cm2,FEHCOO-为98.4%,高于电还原BiOBr0.5I0.5和BiOCl0.5I0.5得到的纳米Bi(平均尺寸分别为19.5 nm和31.6 nm)的催化性能。
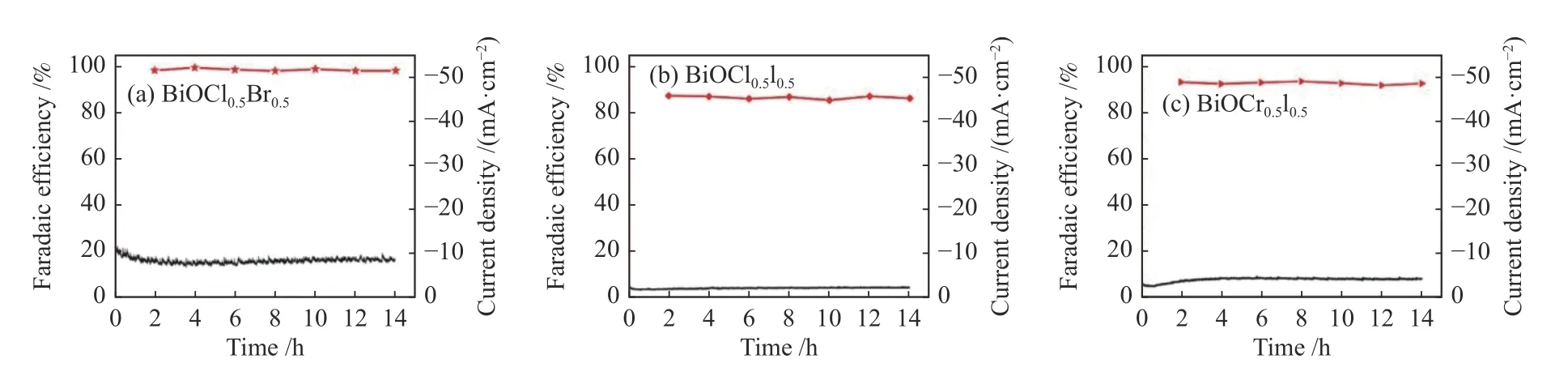
图12 在-1.6 V vs.RHE 电位下对(a)BiOCl0.5Br0.5,(b)BiOCl0.5I0.5,(c)BiOBr0.5I0.5 进行恒电位电解[89]Figure 12 Controlled potential electrolysis on (a) BiOCl0.5Br0.5,(b) BiOCl0.5I0.5,(c) BiOBr0.5I0.5 at -1.6 V vs.RHE[89](with permission from Elsevier publication)
金属化合物催化剂在CO2-ECR 过程中被还原成金属原子,并在催化剂表面发生迁移后又沉积到催化剂表面,由此可以实现金属高指数晶面的有效合成。Yang 等[90]通过DFT 模拟Bi(012)、(111)和(101)晶面的CO2-ECR 过程,发现三个平面都有利于OCHO*的形成。Wei 等[91]通过还原Bi2O2CO3纳米片,在电极材料表面有目的的构筑Bi(012)晶面。DFT 结果如图13 所示,富电子的Bi(012)晶面可以增强CO2分子的吸附,且OCHO*在Bi(012)晶面上的吸附需克服的能垒较低,有利于甲酸盐的生成。

图13 Bi(012)和Bi(003)晶面上OCHO*形成的自由能[91]Figure 13 Free energy for OCHO* generation on (012) and(003) facet of Bi[91](with permission from Elsevier publication)
除了调控催化剂的形貌、尺寸和晶面,在催化剂表面有目的地进行掺杂,改变局部结构并引入缺陷,也是提高反应性能的有效手段。Zhang 等[92]通过水热和氨化过程使氨分子的N 原子取代氧化铟纳米片的O 原子,并在体系中留下氧空位,以此制备了一种同时引入N 掺杂剂和氧空位的多孔氮氧化铟纳米片。电化学结果和DFT 显示,OCHO*的形成是决速步骤,N 掺杂和氧空位之间的协同作用有助于CO2的活化,加速电子从催化剂表面向反应中间体的转移,并降低OCHO*氢化生成甲酸盐的反应自由能。在H 型电解池(电解液为0.5 mol/L KHCO3)和-0.8 V vs.RHE 电位下,甲酸盐的选择性达到95.1%;在流动池(电解液为1 mol/L KOH)和-1.13 V vs.RHE 电位下,甲酸盐部分电流密度可达121.1 mA/cm2。
3.2 电解液
电解液也是影响CO2电还原性能的一个重要因素,其在电还原过程中与电极表面共同提供质子/电子交换场所、构筑催化剂表面微环境、将CO2分子输送到电极表面。目前,实验室研究中广泛使用的是水系电解液,根据其pH 值可划分为酸性、中性和碱性电解液。在酸性体系中,CO2直接与H+结合替代CO2与H2O 反应,形成OCHO*中间体,提高CO2电还原速率,同时酸性体系还能避免碳酸盐/碳酸氢盐的形成,从而提高碳利用率。HER 是CO2-ECR 过程最具竞争性的副反应,尤其在酸性电解液中,H+更容易迁移到电极表面,占据CO2-ECR 活性位点,从而导致更剧烈的HER。Jouny 等[38]在酸性电解液中探究了CO2-ECR 性能,研究发现,在pH=0.9 的H2SO4中进行电化学测试时,所有电位下的FEHCOO-均小于10%。当添加KCl后,K+的引入减慢了H+向电极表面的传输,降低了电极表面的质子覆盖率,从而增大CO2分子到活性位点的可达性,FEHCOO-可达到92.2%。为了抑制HER 和实现高效CO2电还原制甲酸,中性和碱性电解液被广泛应用于CO2-ECR,人们普遍认为,电极表面的碱性环境有利于CO2的活化,而中性电解液由于CO2-ECR 的进行和由HER 导致的质子损耗,导致电极表面局部pH 值升高,与碱性电解液在催化剂表面构筑的微环境类似,因此,中性电解液也有利于CO2活化。
电解液的阴、阳离子虽然不直接参与CO2-ECR过程,但不同的阴、阳离子对甲酸盐的部分电流密度和法拉第效率的提高具有不同的促进作用。Zhang 等[93]在MHCO3(M=Li+、Na+、K+、Rb+、Cs+)的水溶液中使用SnO2催化CO2电还原生成甲酸盐,如图14(a)所示,反应活性按以下顺序逐步升高:Li+<Na+<K+<Cs+<Rb+。随着阳离子半径增大,甲酸盐的法拉第效率增加,而生成H2和CO 的法拉第效率显著降低。这是因为阳离子在溶液中以水合阳离子形式存在,阳离子的水合数与阳离子半径成反比,这导致半径较大的阳离子(如Cs+、Rb+)水合后的尺寸反而小于半径较小的阳离子(如Li+、Na+、K+),在电极表面更密集,促使电极表面电荷密度增大,有利于CO2的吸附,但由于电极表面对水合阳离子的过度吸附,导致可用于吸附活化CO2的位点数减少,因此,CsHCO3电解液中CO2-ECR 性能略差于RbHCO3电解液中的CO2-ECR 性能。

图14 (a)电解质类型和阳离子尺寸对电流密度和FEHCOOH 的影响 0.1 mol/L 的MHCO3(M=Li +、Na +、K +、Rb +、Cs + )电解液,-1.4 V vs.RHE 电位,SnO2/C 电极;(b)0.1 mol/L 的NaX 和KX(X=H C、Cl-、Br- and I-)电解液中,-1.4 V vs.RHE 电位下,SnO2/C 电极上得到的电流密度和FEHCOOH[93]Figure 14 (a) Current density and FEHCOOH as functions of electrolyte type and the cation size,measured using an SnO2/C electrode at -1.4 V in 0.1 mol/L MHCO3 (M=Li +,Na +,K +,Rb +,Cs +) electrolytes;(b) current density and FEHCOOH at the SnO2/C electrode at -1.4 V in 0.1 mol/L NaX and KX (X=HC,Cl-,Br- and I-),respectively[93](with permission form Elsevier publication)
在以往的研究中,抑制HER、提高产物选择性一直是研究的重点。一般来说,水、质子、碳酸氢盐、各种质子化合物等都是CO2-ECR 过程中的质子来源。Chen 等[94]通过原位红外吸收光谱证实水是主要的质子来源,并针对抑制H+形成和HER 提出了不一样的看法,认为催化剂界面对质子的亲和力在决定CO2-ECR 的活性和选择性方面有重要影响。例如在FeN4位点附近掺杂硫,产生用于水解离的活性位点以促进质子生成,与催化剂表面的活化CO2耦合,可以有效提升CO2-ECR 性能。DFT 也证实了经质子转移动力学调整的CO2-ECR过程的反应能垒低于单一FeN4位点上的反应能垒。基于此,Chen 等[95]在进一步的工作中通过在Bi2O2CO3上构建具有强的氧亲和力的氧空位,从而与水发生快速相互作用,促进水的活化,调整质子转移动力学,实现高效CO2-ECR 性能。
3.3 杂质气氛和反应器
在CO2-ECR 过程中,阴极电解液中的金属离子杂质会发生电化学还原,沉积在电极表面,覆盖催化剂活性位点,降低电还原性能[96]。此外,反应气氛中的杂质气体也会显著影响CO2-ECR 过程。在实验室研究中,所用的CO2气体相对于实际生产排放的CO2更为纯净,因此,鲜少有研究关注到杂质气氛是如何影响CO2还原的。Luc 等[97]在近中性电解液中研究了SO2分别对Ag、Sn、Cu 催化剂上CO2-ECR 的影响,如图15 所示。在热力学上SO2比CO2更易还原,且更容易占据电极表面活性位点,对于Ag 和Sn 催化剂上的电还原过程,通入SO2和CO2混合气时,CO2-ECR 的产物总法拉第效率下降;但当停止通入SO2时,CO2-ECR 的性能恢复,表明SO2对CO2-ECR 的影响是可逆的。对于Cu 催化剂,在通入SO2和CO2混合气体时,CO2-ECR 产物的总法拉第效率下降,且CO2-ECR 的主产物从CO 转换为甲酸盐,C2+产物的生成被抑制,说明反应路径发生转变。停止通入SO2,甲酸盐的生成仍占主导,CO 的生成逐渐增加。CO*二聚是C2+产物生成的前提,因此,随着SO2停止通入,C2+的生成逐渐增多。对于实际的工业生产,CO2电还原过程的气体组分更为复杂,因此,有必要进一步深入研究其他杂质气氛(如NOx、CO 等)对CO2电还原活性和选择性的影响。
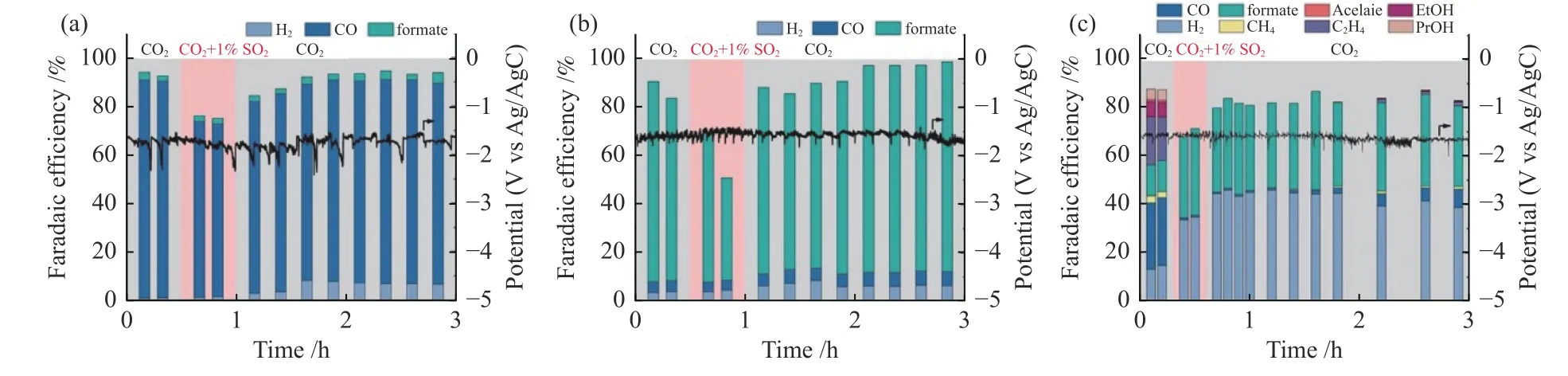
图15 在1 mol/L KHCO3 中100 mA/cm2 的恒定电流密度下,在(a)Ag,(b)Sn 和(c)Cu 催化剂上的CO2+SO2 催化性能[97]Figure 15 Performance of CO2+SO2 electrolysis over (a) Ag,(b) Sn and (c) Cu catalysts at constant current density of 100 mA/cm2 in 1 mol/L KHCO3[97](with permission from ACS publication)
除了上述几种影响因素外,反应器也会影响CO2-ECR 性能,其本质是减小装置内阻,改善CO2传质来提升CO2电催化还原性能。目前,应用最普遍的是H 型电解池。在H 型电解池中,CO2通过溶解和扩散的方式到达电极表面,但由于CO2在水系电解液中的溶解度很小,严重限制了其转化效率,导致H 型电解池中的CO2-ECR 性能无法达到工业化生产的要求。基于此,研究者开发了配备疏水性气体扩散电极的流动池以解决CO2的传质问题。Qian 等[98]合成了纳米棒负载二维纳米片的SnO 电催化剂,然后分别在含有0.5 mol/L KHCO3溶液的H 型电解池和含有1 mol/L KOH 溶液的流动池中进行了电还原CO2生成甲酸盐的性能研究。结果显示,在H 型电解池中,CO2生成甲酸盐的最大电流密度为60 mA/cm2,并且是在较负的电位下实现的;而在流动池中,在-0.7 V vs.RHE时,甲酸盐的部分电流密度可达到330 mA/cm2,甲酸盐的FE 达到94%,CO2电还原生成甲酸盐的能量转换效率为70%,同时由于CO2传质限制的改善和CO2吸附浓度的提高,在-0.2 V vs.RHE 电位时就检测到有甲酸盐生成。该结果说明流动池中电还原CO2性能优于H 型电解池,这主要是因为在流动池中,CO2不与电解液接触,被直接输送到催化剂表面,更有利于CO2的电还原,但CO2易穿过气体扩散电极与另一侧的电解液发生反应,生成碳酸盐/碳酸氢盐沉积,导致电解质数量减少,CO2利用率降低。Xia 等[99]开发了固体电解质(Solid-state electrolyte,SSE)电解池,用以改善碳酸盐/碳酸氢盐沉积问题,实现CO2向纯HCOOH 的连续电催化转化。在这种固体电解质电解池中,可以实现电化学产生的阴离子(HCOO-)和阳离子(H+)直接结合生成纯产物(HCOOH),并提供有效分离CO2还原产物(HCOOH)的通道,降低产品分离过程的成本。
4 总结与展望
本工作对近五年来CO2电化学还原生成甲酸盐的研究进行了综述总结,从催化反应机理出发,聚焦目前各种类型催化剂的特点及反应性能主要影响因素,旨在探讨如何通过催化剂和反应体系的设计来提高CO2-ECR 性能,以促进该领域的研究和发展。近年来,CO2电化学还原制备甲酸盐领域的发展迅速,实验室规模的研究已实现超过90%的甲酸盐法拉第效率和高达1.9 A/cm2的甲酸盐部分电流密度,但仍存在许多问题限制了其工业化应用,例如如何提高CO2的单程转化率,如何在长期运行的过程中维持高稳定性,以及后续的甲酸盐分离纯化等。目前,研究的重点仍在于实现优异的催化性能,这离不开催化剂、电解液和反应器的共同进步。结合现有的研究成果,提出以下研究方向:
第一,新型高效催化剂的设计与合成。一方面持续深入研究金属催化剂,尤其是对纳米材料和复合材料的创新,如设计开发新的合成策略,改变元素种类和组合,合成具有三维结构的高熵合金催化剂;另一方面,设计合成同时适用于阴极CO2-ECR 和阳极电氧化的双功能催化剂,从而实现采用单一的催化剂即可实现甲酸盐的高效的工业级电合成。
第二,原位表征和理论计算相结合,深入探究CO2电还原催化过程本质。高效催化剂的理性设计源于对电化学界面过程和催化剂表界面结构的研究。将电化学组件和各种原位表征技术结合,探究反应过程中催化剂表面活性中间体的动态演变,并辅以DFT 理论计算来阐明实际活性位点及其反应机理,将是未来研究的一大趋势。
第三,新型电解液体系的设计开发。目前,所使用的电解液主要分为水系和非水介质,其中,水系电解液的应用最为广泛,但存在目标产物的选择性低,以及伴随整个还原过程的严重的竞争性析氢反应等问题。非水介质(如离子液体等)的高成本和需要在高电位下电还原CO2限制了其大规模应用。因此,有必要开发更多的新型电解液体系,例如固态电解质等,以满足提高CO2溶解度、降低还原电位和抑制HER 的需要。
第四,新型电解池的设计开发。尽管目前已经有很多电催化剂实现超过90%的FEHCOO-,但在实际的电解过程中,CO2的投入都明显过量,且甲酸盐易穿过离子交换膜到达阳极从而被氧化,导致了CO2的单程转化率非常低。因此,仍需对电解池进行优化,例如改进气体扩散电极、优化离子交换膜等。设计新型电解池提高CO2单程转化率并得到高浓度的甲酸盐产品,仍是广大研究者们面临的难题。
