建立并验证25个祖源信息标记物的集合用于族群信息推断
2023-11-13李耀庭
李耀庭
(广东警官学院刑事技术系 广东 广州 510440)
1 引言
单核苷酸多态性(Single Nucleotide Polymorphisms,SNPs)是指基因组中特定位点的单个碱基变异形成的多态性,绝大多数SNPs呈现基因座上二等位基因的变化。SNPs具有在人类基因组中分布广泛(高于1%的人群比例)、突变率低、较高的遗传稳定性等特征,适合对各个族群间的生物特征进行区分。在各个族群之间频率分布差异较大的SNPs被称为祖源信息标志物(Ancestry Informative Markers, AIMs)。传统的法医DNA鉴定主要基于比较供试者与参照样本的STR(Short Tandem Repeat,短串联重复)位点的一致性。相比STR,SNPs位点具有在基因组上分布广、低突变率、序列短、犯罪现场的高度降解样本也能被检测等优点,有望成为刑事物证鉴定手段之一[1-2]。
随着高通量测序技术的不断成熟,测序成本和数据分析门槛大大降低,使高通量测序技术应用在法医DNA分析领域成为研究热点之一。在法医DNA检测中,STR因具有高度的多态性而成为最传统的检测目标,适合进行人类身份鉴定。但高通量测序技术由于可以区分具有相同长度但不同序列的等位基因,从而允许对等距离等位基因进行全面分析,提供了比给定等位基因重复次数更多的信息。但高通量测序数据在STR进行基因分型准确率方面仍有不足,因为许多STR等位基因长度过长导致测序误差率很高,需要提高测序覆盖率才能获得比较可靠结果。此外,由于测序读长大小的限制,难以检测较长等位基因,会引起侧翼区域突变导致空等位基因突变。
SNP是DNA序列中单个碱基的改变。在人类基因组中,SNP是最常见的标记,迄今已经发现了超过8400万个SNPs位点。这些标记已广泛用于法医学研究,因为SNP的PCR引物和测序读长较短,可以提高对降解样本成功检测的可能性。随着高通量测序平台的应用,大规模进行SNPs分析成为现实,例如千人基因组计划、人类基因组多样性项目和Simons多样性计划等,这些大规模的高通量测序项目使得近3年注释了数百万新SNPs位点,并提供了全球SNP等位基因频率的数据集。高通量测序技术一次能获得百万级别的SNPs信息,可为个体识别和表型推断提供大量生物特征信息,为案件侦查指明更为详细的办案方向[3]。基于AIMs研究及其反映的生物地理学的祖源信息,已成为法庭科学界的研究热点[4]。目前已报道了一些用于族群区分的AIMs集合,如Phillip等[5]报道常染色体34个SNPs集合能有效区分三大洲际(非洲,东亚和欧洲)人群,该项研究结果在11-M马德里爆炸案调查中得到了应用,并提供了有关负责恐怖组织的重要线索。随后,Phillip等对SNPs集合进行优化和调整,扩大范围覆盖至五大洲际人群[6]。Kidd等[7]建立55个AIMs的集合用于对五大洲际族群及其亚群进行划分,Kidd随后构建74个AIMs的集合[8],增加了对亚洲人群的划分。随着高通量测序技术的成熟,越来越多高精度和高灵敏度的AIMs被报道。这些AIMs在检测低含量的DNA样本中有着更好的商业应用前景。如Kidd 的集合也被加入到Verogen公司生产的ForenSeq;Thermo Fisher Scientific公司[9]建立165个常染色体的AIMs集合,该集合能有效区分多个主要族群。
然而,当前的AIMs集合普遍检测位点较多,需要具有生物实验和大数据分析背景的专业人才进行研究,难以满足公安实战简便检测、快速检测和微量样本检测的需求。因此,本研究以千人基因组计划的公开数据[10],基于朴素贝叶斯分类器的人工智能算法,从8000万个SNPs中挑选25个高信息密度的AIMs组成集合(Li-25AIMset)。评估和验证Li-25AIMset在训练集和测试集中对未知样本的祖源信息的预测能力,分析族群推断模型在公安实战的应用价值。
2 材料与方法
2.1 数据样本整理
本研究所用的数据共2504个样本的全基因组SNPs,如表1所示,包括非洲样本661份,混合美洲样本347份,东亚样本504份,欧洲样本503份,南亚样本489份。将样本数量按照80%∶20%分为训练集和测试集。
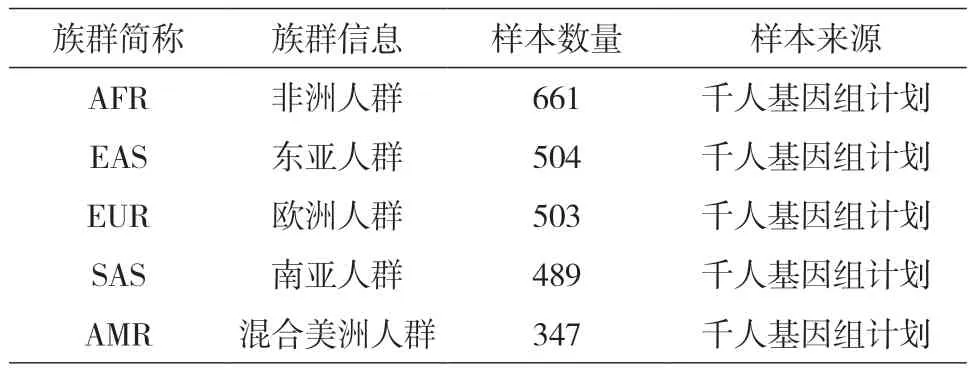
表1 千人基因组数据集样本信息
2.2 双等位基因SNPs过滤和整理
使用vcftools(Adam Auton等[11])对SNPs数据集的每个染色体的SNPs数据进行过滤,提取双等位基因SNPs并保存为biallelic.vcf.gz文件。利用vcftools提取数据集中SNPs等位基因突变频率,计算双等位基因SNPs在各种人群突变频率的信息量度值(Informativeness,In),In值反映SNPs在不同人群中突变频率的差异[12-13],选择90%分位数的In值作为筛选阈值,大于阈值的SNPs进行后续分析。
2.3 朴素贝叶斯分类器算法筛选SNPs
以朴素贝叶斯分类器算法计算SNPs对样本所属于类别的概率[14-15]。该算法是一种基于概率的分类方法,即假设特征之间相互独立,计算该特征属于某一类别的概率,并选择概率最大的类别作为预测结果。使用对数损失值(logarithmic loss,logloss)评估预测准确性,公式为:
选择对数损失值最小的SNPs作为候选的AIMs。
2.4 AIMs推断能力的评价方法
在公安实战应用中,提供相同的信息量且包含更少位点的SNPs集合更具有应用前景。为实现这个目标,需要剔除信息量较少或冗余的SNPs以获得最精简的体系,并评估模型预测的准确度。使用vcftools对AIMs计算成对固定指数(Wright's fixation index,Fst)。Fst是衡量族群中基因型实际频率是否偏离遗传平衡的常用指标。Fst取值范围为(0,1),数值越大,表明等位基因在各自人群中越固定(频率越高),群体间分化程度越大。使用R语言(Rstudio, Posit公司)的pheatmap包绘制非洲、东亚、欧洲和南亚人群AIMs等位基因频率的热图。使用Plink程序[16]、Python语言(Jupyter Notebook,Jupyter公司)的UMAP(Uniform Manifold Approximation and Projection)[17],以等位基因频率进行分析,以降维形式评估族群之间的距离。使用fastSTRUCTURE程序[18]基于AIMs进行的族群聚类分析,以非监督聚类方式评价族群成分的数量,探索大陆族群的遗传结构。使用测试集数据评价AIMs集合对未知样本的预测性能。
3 实验结果
3.1 千人基因组数据集数据整理
本研究基于千人基因组计划—第三阶段的公开数据集,其中包含5个族群的共2504个样本的基因分型数据集。将样本按照80%∶20%分为训练集和测试集,数据集共包含大于8000万个SNPs的位点。混合美洲人群因为其样本混合其他大陆的祖先背景,不能反映该族群的生物特征性[19],故在后续的分析中排除。
3.2 朴素贝叶斯分类器算法挑选潜在的祖源信息标记物(AIMs)
使用朴素贝叶斯分类器算法,在训练集样本中筛选潜在的AIMs,使用logloss值对不同数目AIMs集合的族群推断能力进行评价。使用AIMs集合对训练集数据的族群信息进行推断,并绘制AIMs数量在训练集中logloss值的折线图,如图1所示。随着集合中AIMs数目增加,logloss值逐渐减少,表明对未知样本的祖源判断为错误族群的概率在不断减少。当AIMs数量大于15,logloss值接近0,表明该模型能准确判断训练集中样本的族群信息。
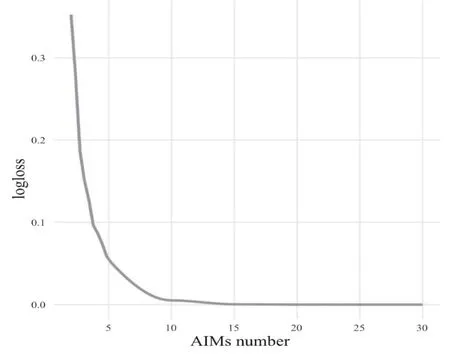
图1 族群信息推断集合AIMs 的数量在训练集中logloss 值的折线图
经过朴素贝叶斯分类器选择的30个候选AIMs的详细信息,包括Rs ID号码,位点所在的染色体,所在染色体位置,参考核苷酸(REF)、突变核苷酸(ALT)、Fst值,如表2所示。Fst数值越大,表明等位基因在各自族群中越固定,群体间分化程度越大。表2列出AIMs在各族群间的Fst值,平均值为0.46,所有Fst均大于0.15,表明挑选的AIMs能较好地区分各个族群。

表2 30 个候选祖源信息标志物的详细信息
3.3 测试集数据评价AIMs集合预测能力
使用测试集数据对AIMs模型的族群预测能力进行验证,通过logloss值对不同数目AIMs集合的预测能力进行评估,并绘制AIMs数量在测试集中logloss值的折线图,如图2所示。随着AIMs数目增加,logloss值逐渐减少,当AIMs数量大于等于25,logloss值接近0,表明AIMs的数量大于等于25个,能成功地预测所有未知样本。此后继续增加AIMs的数量,logloss值变化微乎其微。
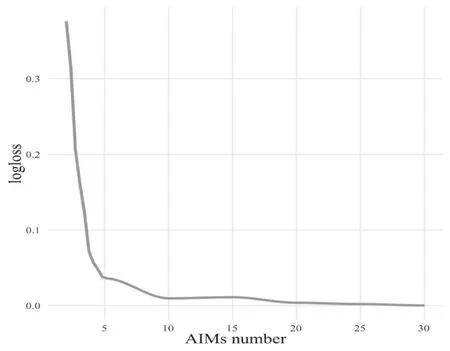
图2 族群信息推断集合AIMs 数量在测试集中logloss 值的折线图
3.4 Li-25AIMset在不同群体突变频率分析
基于上述分析结果,建立筛选出的前25个AIMs模型,命名为Li-25AIMset,并对该模型的预测能力进行评价。以Li-25AIMset在四个族群中等位基因频率绘制热图,如图3所示。颜色的深浅代表了该AIMs位点的基因频率在不同群体中的相似性和差异性,颜色越深表示该AIMs位点在对应族群的基因频率越高,越浅表示该位点在对应族群的基因频率越低。通过AIMs在各个族群所呈现颜色的深浅,能明显区分各个族群。此外,热图上方的聚类树将具有相似分布频率的AIMs聚集在相同分枝。例如,rs6064851、rs1436148、rs2814778、rs8033972被识别为非洲人群特征信息位点;rs16891982、rs1148596、rs12881545被识别为欧洲人群特征信息位点;rs922452、rs12920407、rs10216541、rs10434693、rs1906496被识别为东亚人群特征信息位点。
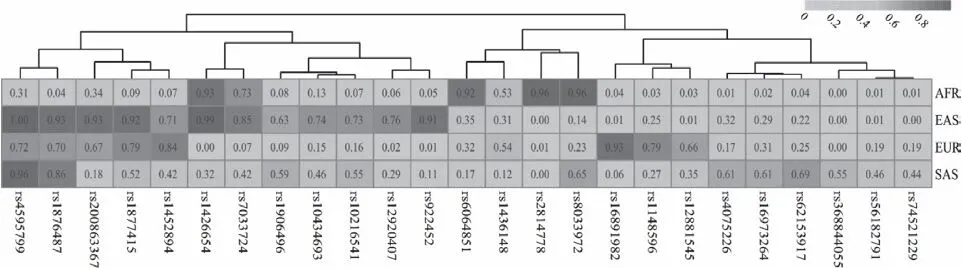
图3 25 个祖源信息标志物集合(Li-25AIMset)在各个族群中等位基因频率热图
3.5 Li-25AIMset群体遗传结构分析
UMAP降维分析可反映个体水平上的族群结构,不同的颜色代表不同的族群,样本的聚类情况可反映族群间的遗传关系,同时根据不同的距离可进行族群的区分。基于Li-25AIMset的UMAP分析结果如图4所示,Li-25AIMset能将相同族群的样本形成集落,并形成4个聚类集落,对不同族群的样本明显区分,在图中以4种序号标记表示。
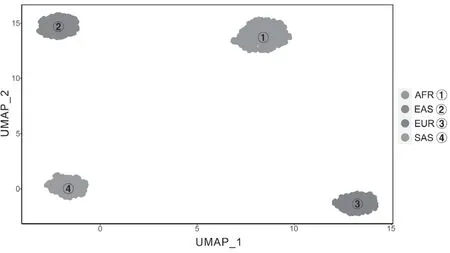
图4 基于Li-25AIMset 在各个族群的祖先起源UMAP 分析
基于Li-25AIMset的STRUCTURE聚类分析在K=2,3,4,5时的结果,如图5所示。个体水平祖源成分见柱状图,其中每个柱形表示为一个样本,每种颜色代表一个祖源成分,每个竖条中各颜色的长度对应各个样本的祖源成分比例。随着K增大,族群间的区分程度越明显。当K=2时,非洲族群表现出高红色占比,并与其他族群区分;当K=3时,东亚和非洲族群能与其他族群区别;当K=4时,可观察到非洲、南亚、东亚和欧洲族群间具有明显的遗传差异,呈现明显的集落。当K继续增大时,族群间并没有进一步的划分。图5结果表明Li-25AIMset能清晰地区分非洲、欧洲、南亚和东亚人群之间存在差异性。

图5 基于Li-25AIMset 在不同人群的祖源STRUCTURE 分析
3.6 评价Li-25AIMset模型
基于测试集数据(2 0%样本),对比L i-25AIMset和已发表的祖源SNPs集合(55Kidd-AIMset)进行族群信息预测,对预测结果进行评价和比较,评价指标是灵敏度(Sensitivity)、特异性(Specificity)、阳性预测值(PPV)、阴性预测值(NPV)、Kappa系数(10折交叉验证)、准确率(95%置信区间)、logloss值等参数,如表3所示。两个模型在测试集数据的预测准确率均达到100%。但是,Li-25AIMset在数据中的10折交叉验证的平均Kappa系数为99.94%,大于55Kidd-AIMset的99.05%。此外,Li-25AIMset的logloss值(4.8×10-7)明显低于55Kidd-AIMset(4.2×10-2),预测为错误族群的可能性显著更低,并且检测位点数目更少,位点数目仅为55Kidd-AIMset集合的45%。

表3 Li-25AIMset 与55Kidd-AIMset 模型预测能力结果对比
4 讨论与结论
4.1 朴素贝叶斯算法在基因信息祖源推断中的应用
使用生物标志物进行祖源推断在证据调查中发挥着愈发重要的作用。随着基因分型和高通量测序技术的发展,基于基因信息的祖源推断技术有望应用于案件侦查。然而在实际应用中,现场样本通常是微量并且降解的,急需简易操作、检测标的少,且能提供足够有效生物特征信息的技术。本研究利用朴素贝叶斯分类器算法对千人基因组计划全基因组SNPs数据进行了训练,建立了Li-25AIMset族群推断模型并对模型的预测能力进行验证。从研究结果来看,Li-25AIMset能成功地将训练集和测试集数据判断为正确的族群。基于Fst值、UMAP图和STRUCTURE图,表明Li-25AIMset模型能反映出四个族群显著不同的生物特征。
在筛选潜在的AIMs方面,朴素贝叶斯分类器相比其他算法,其只需小规模的数据量就可以让算法有较高的训练准确率。此外,该算法能较好地处理较多分类项目,适合增量式训练。随着样本量的逐渐增大,朴素贝叶斯分类器算法会逐渐变得比决策树算法更快,在大数量训练和查询时具有较高的速度。此外,该算法计算速度远远胜过支持向量机算法,收敛速度快于判别模型。该算法基于条件独立性假设,对样本的类别进行相互独立判断,适合于解决对未知样本判断属于已知类别的问题,STRUCTURE和SNIPPER等软件都是基于该算法进行SNPs数据分析。如Cheung等[20]通过比较朴素贝叶斯分类器、遗传距离和多项式逻辑回归算法,发现朴素贝叶斯分类器算法预测的准确率更高。本研究结合朴素贝叶斯分类器的优点,使用该算法筛选候选的AIMs,在千人基因组计划数据集对族群信息推断上取得了较好的结果。
4.2 法医DNA表型学及其在犯罪个体画像中的应用
近5年来,法医DNA表型学备受法庭科学学界关注。该领域旨在通过分析DNA变异(主要是SNPs特异性),以预测个人的外观特征(如眼睛颜色、头发形状或雀斑是否存在等特征)。其研究的主要目标是当调查陷入僵局时,通过预测外观特征可以为警方提供额外线索。然而,外观特征是复杂多基因性状,有数百个基因参与其中。
本研究建立了Li-25AIMset模型,通过heatmap结果,发现rs922452、rs12920407、rs10216541、rs10434693、rs1906496的位点能反映东亚人群特征信息。其中rs922452位于EDAR基因,与头发的特征,如厚度和平直度等相关[21]。此外,结果显示rs6064851、rs1436148、rs2814778、 rs8033972是非洲人群特征信息位点。其中rs2814778在DARC基因的位置,与非洲种族中性粒细胞减少症的相关[22],如Legge等[23]发现rs2814778与氯氮平治疗引起中性粒细胞减少相关,并且存在非洲祖源的特异性。此外,rs16891982、rs1148596、rs12881545被识别为欧洲人群特征信息位点,如Branicki等[24]报道rs16891982位于SLC45A2基因,与毛发颜色有关,并在欧洲族群富集。而rs1426654位点在SLC45A2基因的不同区域,在南亚族群富集,如Wilde等[25]报道rs16891982参与影响皮肤,头发和眼睛色素呈现,并推断欧洲人种最近5000年的变化轨迹。此外,rs16891982还与黑色素瘤、基底细胞癌的易感性相关。基于本研究发现的特征性AIMs及特征性位点所反映的外观特征,将在后续实验中验证其反映的个体外观特性,从而加深对法医DNA表型学的认识,为精准描绘犯罪个体画像提供理论基础。基于此,可开发通过高通量测序技术进行DNA分析外貌、年龄和祖先来源的综合预测工具。
4.3 Li-25AIMset在公安业务实战中的应用前景
通过UMAP和STRUCTURE方法分析,Li-25AIMset集合能准确分辨族源信息,四个族群的区分度显著,表明模型具有较强的分析性能。相比55Kidd-AIMset模型,Li-25AIMset模型准确率更高,logloss值更低,并且检测位点更少。本研究建立的Li-25AIMset模型在保证推断结果的高准确率的前提下,只用更少的检测位点就可提供更多的信息量,有利于在公安业务的实战应用。
总体而言,本研究建立的Li-25AIMset预测模型能实现对非洲族群、欧洲族群、东亚族群和南亚族群的推断。由于本研究所使用的人群数据集来源较单一,且验证集数据量有限,因此,后续需扩大数据来源继续验证模型的稳健性。通过收集其他数据集洲际人群的数据,收集案件现场提取的DNA数据,增加实际测试样本以进一步评估和确定本模型在公安实战中的应用性能,进而提供未知来源样本的族群信息和表型特征等有效线索,为案件侦查指明方向[26]。
