MoX2/MgPSe3(X=S、Se)电子结构与光学性能的理论研究
2023-10-31仇怀利李国军郑雅惠张哲瑞李中军
张 栋, 仇怀利, 李国军, 郑雅惠, 张哲瑞, 李中军
(合肥工业大学 物理学院,安徽 合肥 230601)
自石墨烯得以在实验中制备以来,二维半导体成为了各研究领域的热门材料,其中二维过渡金属双卤族化合物(2D TMDCs)如双卤化钼(MoX2,X=S、Se)凭借其优异的性能,在量子物理、电子材料、光电材料和光催化等领域获得广泛应用[1-3]。近年来,有研究者利用化学气象沉积(chemical vapor deposition,CVD)技术在单层MoS2或MoSe2膜上取代硫族元素,从而成功制备了二维MoSSe薄膜,进一步拓宽了TMDCs在实验方面的应用[4-5]。
在光催化领域,构建半导体异质结是提高光催化活性的一种有效策略[6]。由2种不同的半导体构成的异质结根据能带的相对位置可以分为3种类型,其中能带呈交错排列(Ⅱ型)的异质结对于促进载流子有效分离具有重要作用[7-9]。文献[10]通过第一性原理计算了MoSSe/WSeTe异质结的光催化能力,该异质结具有明显的Ⅱ型能带结构,并且通过空位来增加活性位点提升了该异质结的光催化析氢效率。与此同时,利用双轴应变、外加电场或缺陷等技术来调节二维半导体材料性能的研究也逐渐增加[11-13]。
二维过渡金属磷硫族化合物(APX3,A=V、 Mn、Fe、Co、Ni、Cd、Mg、Zn;X=S、Se)具有独特的磁性和光学特性,可以与其他二维材料互补;不仅如此,此类材料具有层状结构,在氢存储和锂电池中表现出优异的性能,因此受到广泛的关注[14-15]。但是过渡金属磷化硫族化合物普遍具有的宽带隙(1.77~3.94 eV)导致其对太阳光尤其是可见光的吸收能力弱,且其与金属硫族化合物构成异质结方面的研究较少[14]。
本研究采用密度泛函理论计算,通过将MgPSe3与MoS2、MoSe2、MoSSe等二维半导体材料构成异质结,对其电子结构和光学性能进行分析,并且研究缺陷和双轴应变对能带结构的影响。
1 计算模型和方法
1.1 计算模型
通常情况下,MgPSe3、MoS2、MoSe2、MoSSe属于六方晶系,且具有二维层状结构。将MoX2原胞进行2×2×1扩展得到含有12个原子的超胞,并与MgPSe3单胞形成异质结。在研究异质结之前,首先研究单层的MgPSe3和MoX2的晶格常数a、b和带隙Eg,计算结果见表1所列。

表1 二维MoX2和MgPSe3的晶格常数和带隙
计算得到MgPSe3、MoS2、MoSe2、MoSSe的晶格常数和带隙大小与相关文献报道基本一致,且它们的晶格失配率分别为1.5%、2.2%、1.0%,说明两者之间的适配性非常好。
在MoX2/MgPSe3的层间界面处,本文考虑3种对接方式,其俯视图和侧视图如图1所示,分别为X2-Mg、X2-P、X2-Se。因为MoSSe具有面外非对称性,所以针对MoSSe/MgPSe3考虑了2种堆叠结构,分别为MoSSe/MgPSe3_Ⅰ和MoSSe/MgPSe3_Ⅱ,前者MoSSe层中的Se原子向外,后者向内。
为了进一步选取稳定的结构,本文对各异质结的不同结构进行了形成能的计算,计算过程如下:
Ef=EMoX2/MgPSe3-EMoX2-EMgPSe3
(1)
其中:EMoX2/MgPSe3、EMoX2、EMgPSe3分别为MoX2/MgPSe3异质结和独立的单层MoX2、MgPSe3完全驰豫后的总能量。计算结果见表2所列,由表2可知,形成能越小,结构越稳定。

表2 二维MoX2/MgPSe3的形成能Ef
从表2可以看出,无论是哪种异质结,第3种对接方式的形成能都是最大的,证明这种对接方式最稳定。因此之后的计算都是基于这种对接方式进行的。
1.2 计算方法
本文所有计算都基于密度泛函理论的投影缀加波(projected augmented wave,PAW)方法,选用VASP (Vienna abinitio simulation package)软件包完成[21],电子之间的交换相关电势使用带有范德瓦尔斯修正的广义梯度近似下的梯度修正函数(perdew burke ernzerhof,PBE)算法处理。自洽计算和电子结构计算时,布里渊区分别取样为7×7×1和9×9×1。离子驰豫能量收敛标准为 10-5eV,原子间作用力收敛标准为 0.001 eV/nm,并确保计算结果达到收敛标准, 从而得到最稳定的结构。计算过程中的平面波基组截断能量均取为 500 eV。
2 结果分析
2.1 电子学结构
由PBE算法计算得到的MoX2/MgPSe3投影能带及态密度如图2所示。

图2 MoX2/MgPSe3的投影能带及态密度
图2中MoS2/MgPSe3和MoSSe/MgPSe3_Ⅰ是带隙值分别为1.72 eV和1.65 eV的间接带隙半导体,而MoSe2/MgPSe3和MoSSe/MgPSe3_Ⅱ是带隙值分别为1.24 eV和1.32 eV直接带隙半导体。
异质结的直接带隙使得电子的跃迁更加容易,可以有效地提高光吸收效率。
总体上可以看出,当层间界面处为S-Se和Se-Se结构时,异质结分别对应为间接带隙半导体和直接带隙半导体,且随着MoX2中Se成分的增加,异质结的带隙逐渐减小。此外,MoSe2/MgPSe3和MoSSe/MgPSe3_Ⅱ具有明显的Ⅱ型能带结构,根据态密度可以看出异质结的价带顶(valence band maximum,VBM)主要由MoS2贡献,而导带底(conduction band minimum,CBM)主要由MgPSe3贡献。
为了进一步探讨MoSSe/MgPSe3_Ⅱ在光催化析氢方面的应用,本文使用HSE06算法对其电子结构进行计算。该异质结的VBM、CBM经计算分别为-4.23 、-6.26 eV,与水的氧化还原电势(-4.44、-5.67eV)相比较,证明这种二维材料在光催化水裂解方面有应用潜力。
外加应力可以有效调控二维半导体材料的电子结构及相关性质。本文针对这4种异质结,通过改变晶格参数,施加均一的双轴应力,然后绘制各异质结带隙随晶轴拉伸比变化的曲线图。晶轴拉伸比μ定义如下:
μ=(a-a0)/a0
(2)
其中,a0、a分别为原始晶格和晶格应变之后的晶格常数。
MoX2/MgPSe3异质结中MgPSe3和MoX2的CBM与VBM随晶轴拉伸比的变化如图3所示,由图3a、图3b可知,MoS2和MoSSe在晶轴压缩的状态下,其VBM与MgPSe3基本相同,说明了Se-S这种结构的层间相互作用更强。整体来看,各异质结的带隙随着应力的增加逐渐减小。
MoX2/MgPSe3带隙随晶轴拉伸比的变化如图4所示。由图4可知,外加应力对异质结带隙有明显的线性调控作用,且在带隙达到峰值之后存在直接带隙向间接带隙的转变。这种线性调控作用在压变传感方面有潜在的应用价值。
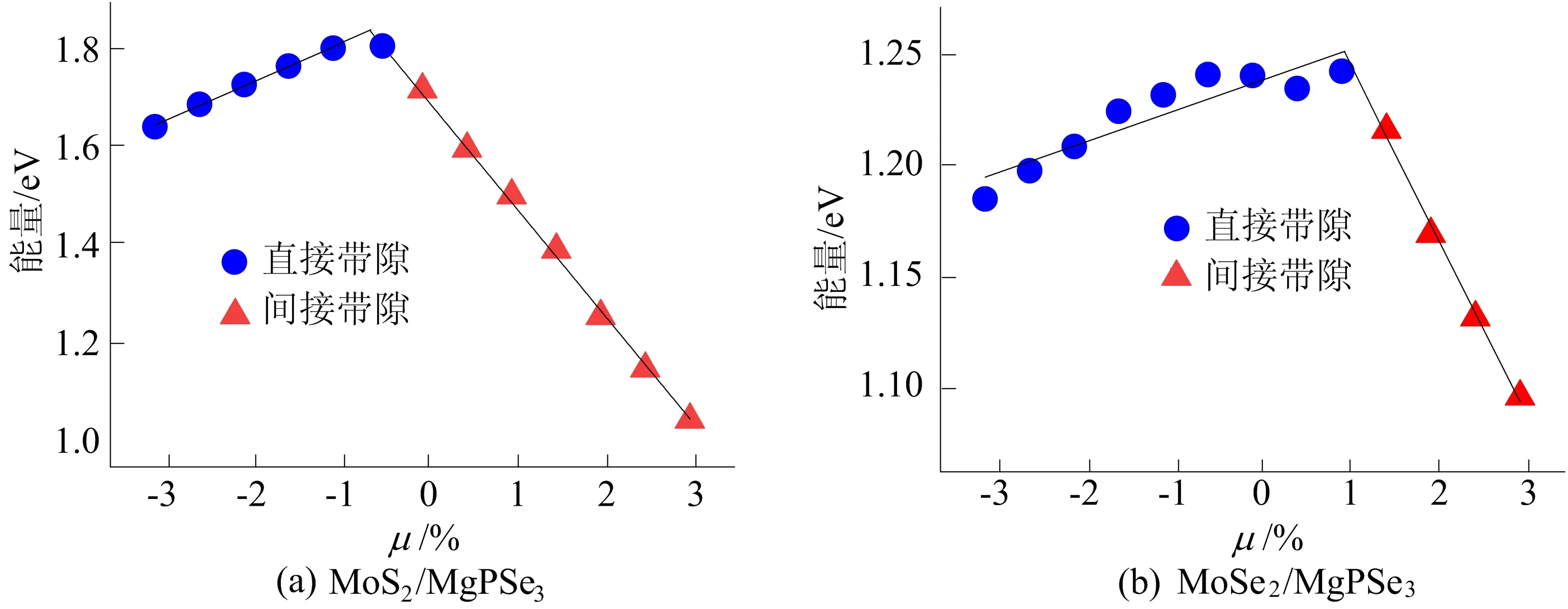
图4 MoX2/MgPSe3带隙随晶轴拉伸比的变化
此外,本文分别在各异质结的MgPSe3层和MoX2层作了缺陷,其结构如图5所示。 MoX2/MgPSe3的空位投影能带图如图6所示,其中左侧为MgPSe3层Se空位,右侧为MoX2层X1空位。

图5 MoX2/MgPSe3中Se空位与X1空位的结构
由图6可知,在异质结的MgPSe3层空位Se之后,其带隙相较原来基本保持不变。事实上,对单层MgPSe3做Se空位,能带结构也基本保持不变,由此可知MgPSe3具有较强的稳定性。此外,在MoX2层空位X1后,能带中出现了明显的缺陷能级,与文献[10]中描述的一致。
2.2 光学性质
随着二维半导体材料在光学领域的不断应用,光吸收系数成为一个衡量其光学性质的重要参数。基于此,本文计算单独的单层MoX2、MgPSe3和MoX2/MgPSe3异质结的光吸收系数。
MoX2/MgPSe3异质结与单层MoX2、MgPSe3的光吸收系数如图7所示,从图7可以看出,相比于单层的MgPSe3,除MoSe2/MgPSe3以外的其他3种异质结的光吸收系数都有明显的提高。其中MoSSe/MgPSe3_Ⅰ和MoSSe/MgPSe3_Ⅱ异质结在紫外光区域的吸收强度有明显的改善,显示其在光吸收方面的良好性能。
3 结 论
本文采用基于密度泛函理论的第一性原理计算方法,研究了MoX2/MgPSe3(X=S、Se)的电子结构和光学性能。计算结果表明,MgPSe3和MoX2的晶格失配率在2.2%以下,且两者界面对接方式为X2-Se时最稳定。通过对MoX2/MgPSe3能带结构的分析发现,MoSSe/MgPSe3_Ⅱ具有明显的Ⅱ型能带结构,且用HSE06算法计算得到的VBM和CBM符合光催化水裂解的要求,证明这种材料在光催化领域具有潜在应用价值。双轴应变对异质结带隙有明显的线性调控作用,且MoS2/MgPSe3和MoSSe/MgPSe32种异质结在晶轴收缩时,有较强的层间交互作用。此外,由光吸收系数的计算结果得到,MoSSe/MgPSe3_Ⅰ和MoSSe/MgPSe3_Ⅱ异质结在紫外光区域的吸收强度比单层的MgPSe3和MoX2有显著的提高,说明异质结对于改善材料的光学性能具有重要作用。
