多基因同步调控结合高通量筛选构建高产L-苯丙氨酸的谷氨酸棒杆菌工程菌株
2023-10-25薛宁王瑾李世新刘叶程海娇张玥毛雨丰王猛
薛宁 王瑾 李世新 刘叶 程海娇 张玥 毛雨丰王猛
(1.天津科技大学生物工程学院,天津 300457;2.中国科学院天津工业生物技术研究所,天津 300308;3.中国科学院天津工业生物技术研究所低碳合成工程生物学重点实验室,天津 300308)
L-苯丙氨酸(L-Phe)在人类和大多数哺乳动物体内不能通过自身合成,是一种必需氨基酸,是重要的食品、化学品及药品中间体[1]。目前,L-Phe可以通过天然蛋白质水解法、化学合成法、酶法和微生物发酵法合成[2-3]。而微生物发酵法由于其对环境友好、原料成本低、反应条件温和、容易大规模培养等优点,成为工业化生产L-Phe的主要方法[4]。
L-Phe的生物合成途径是最复杂的氨基酸合成途径之一,大致可以分为中心碳源代谢途径、莽草酸途径和分支酸途径这3个模块(图1)。目前提高其生物合成的代谢调控策略主要集中在提高前体物质的积累、解除关键酶的反馈抑制作用和转运系统的改造等方面[5-7]。其基因表达调控技术大多依赖于游离型质粒对目标基因(簇)进行过表达,或者常规的基因组编辑技术对不同模块内的多个目标基因进行逐轮迭代改造。但L-Phe代谢途径较长而且调控机制复杂,仅依靠质粒或者周期漫长的逐轮改造[8],难以实现不同模块之间的通量平衡,在一定程度上限制了其产量的进一步提升。
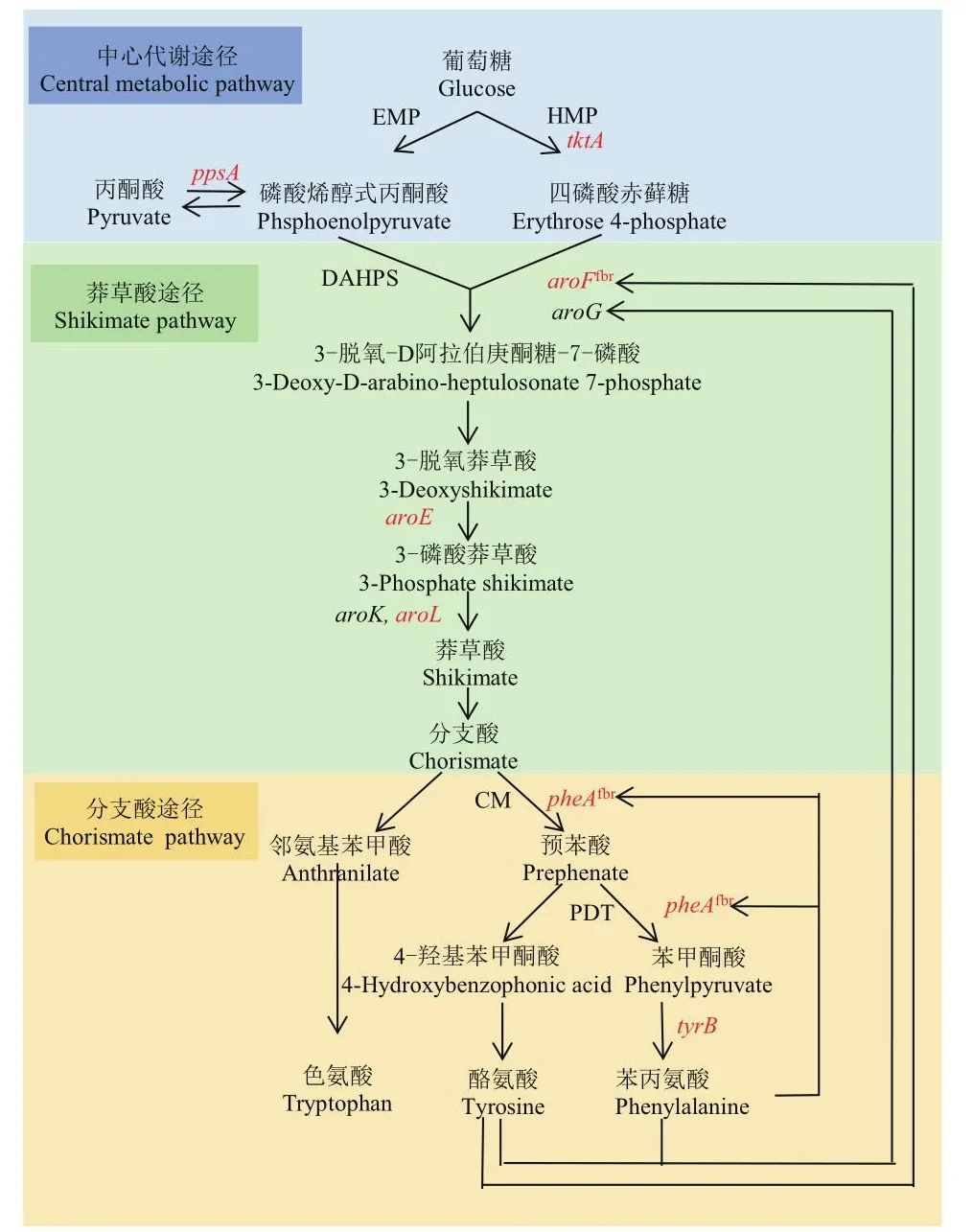
图1 谷氨酸棒杆菌中L-Phe生物合成途径Fig.1 L-phenylalanine biosynthesis pathway in C.glutamicum
目前工业化生产L-Phe的主要菌株为大肠杆菌(Escherichia coli)和谷氨酸棒杆菌,其中谷氨酸棒杆菌具有生物安全、底物谱广泛、营养需求低、基因组编辑技术丰富等优点,是一种理想的微生物生产底盘[9-11]。近年来CRISPR/Cas[12-14]系统介导的基因组编辑技术的发展,极大地丰富了谷氨酸棒杆菌的基因表达调控技术,但基因组上的多基因同步调控方面仍然面临很大挑战。为此,本课题组前期基于CRISPR/Cas系统和胞苷脱氨酶的碱基编辑技术,在谷氨酸棒杆菌中开发了一套多基因表达调控技术BETTER[15]。针对每个基因分别赋予8个G碱基构成的待编辑核糖体结合位点,经过一轮碱基编辑后,成功实现10个基因的同步调控,以色素沉积情况为筛选标准,最优高产番茄红素的菌株获得了2.9 mg/g CDW的产率,优于早先报道的谷氨酸棒杆菌产番茄红素的最高纪录(2.4 mg/g CDW)[16]。该方法使谷氨酸棒杆菌的多基因表达的大规模微调变得简易可行,但后续产品的筛选通量又面临了新的挑战。为此,本课题组近期针对L-Phe开发了一种可在体外使用的基于循环排列荧光蛋白(circularly permuted fluorescent protein, cpFP)的基因编码L-Phe生物传感器[17],既可以应用于胞内反映L-Phe的浓度水平,也可将其纯化在体外直接测定样品中的L-Phe浓度,实现L-Phe浓度的准确测定,其中L-Phe传感器的底物结合域可以特异性结合L-Phe并发生蛋白质构象变化,而对蛋白质构象变化敏感的cpFP可将偶连的蛋白构象变化转变为荧光信号的变化。并与液滴微流控高通量筛选技术结合[18-19],有望实现L-Phe高产菌株的快速筛选。
本研究采用BETTER多基因调控技术,针对谷氨酸棒杆菌中L-Phe生物合成途径的3个模块的7个关键基因ppsA、tktA、aroFfbr、aroE、aroL、pheAfbr和tyrB的RBS进行同步调控,产生大量的不同RBS组合的菌株突变文库,并通过L-Phe生物传感器结合液滴微流控筛选技术,筛选L-Phe合成显著提升的突变株,以期为L-Phe工程育种提供一种可行策略。
1 材料与方法
1.1 材料
1.1.1 菌株和质粒 本研究所用菌株和质粒均为作者实验室保藏和构建,详见表1。

表1 本研究用到的菌株和质粒Table 1 Strains and plasmids used in this study
1.1.2 酶、引物及相关试剂盒 Q5 High-Fidelity DNA聚合酶构自NEB公司,2×Es Taq Master Mix(Dye)购自康为世纪有限公司;引物由北京擎科生物有限公司合成,详见表2。大肠杆菌感受态细胞制备试剂盒购自TaKaRa公司,质粒小量抽提试剂盒、琼脂糖凝胶回收试剂盒、基因组DNA快速抽提试剂盒购自天根生化科技有限公司,重组试剂盒购自南京诺唯赞生物科技有限公司。
1.1.3 培养基 LB培养基(g/L):胰蛋白胨10,氯化钠10,酵母提取物5;BHI 培养基(g/L):BHI 37,(NH4)2SO410,K2HPO40.2,NaH2PO40.3,MgSO4·7H2O 0.5,pH 7.2;BHIS培养基(g/L):山梨醇 91.1,BHI 18.5,pH 7.2;LBHIS培养基(g/L):蛋白胨5,NaCl 5,酵母粉2.5,BHI 18.5,山梨醇91.1,pH 7.2;NCM培养基(g/L):具体配方详见参考文献[20];CGX II培养基(g/L):具体配方详见参考文献[21]。
1.2 方法
1.2.1 质粒构建及转化 对L-Phe生物合成途径的3个模块的7个关键基因ppsA、tktA、aroFfbr、aroE、aroL、pheAfbr和tyrB 作为目标靶点进行遗传改造,如图1所示。本研究用于质粒构建和构建过程的引物序列表详见表2。借助常规分子生物学技术构建pK18-ΔaroP::tktA-ppsA和pK18-ΔldhA::aroE-aroL-tyrB重组质粒,其中aroFfbr、pheAfbr课题组前期已完成整合。此外,为了表征突变后的RBS对于目标基因翻译效率的影响,构建了含有不同RBS元件、不同目标基因前180 bp、Linker(GGGGSGGGGSGGGGS)与绿色荧光蛋白GFP融合的pEC-XK99E系列质粒。谷氨酸棒杆菌的转化及阳性重组子的筛选参考已发表工作[22]。
1.2.2 利用碱基编辑方法产生RBS突变库 将质粒pgRNA-RBS2和pXMJ19-nCas9-AID电转入构建好的底盘菌株感受态中,得到待编辑菌株并以初始OD600=0.2转接至CGX II(20 g/L葡萄糖)中30℃诱导编辑20 h,IPTG浓度为0.5 mmol/L。随后将其转接到LBHIS培养基中在37℃摇床中过夜培养,利用较高温度将温敏型质粒pgRNA-RBS2和pXMJ19-nCas9-AID去除。利用菌落PCR分别扩增整合至ΔCGP2、ΔaroP和ΔldhA位点的人工模块的DNA片段,一代测序验证突变库中的7个基因的RBS编辑结果。
1.2.3 液滴微流控分选
1.2.3.1 液滴生成 将上一节已去除质粒的RBS突变文库转接至CGX II(60 g/L葡萄糖)培养基中并调整OD600=0.1,作为液滴生成第一水相;将体外纯化的L-Phe生物传感器(蛋白)溶于CGX II(60 g/L葡萄糖)培养基作为液滴生成的第二水相;含有表面活性剂008-fluoroSurfactant(2%)的HFE 7500作为油相;调整流速至液滴直径为23 μm左右。收集大约100 μL液滴于30℃培养箱中静置培养,不同时间点取样于荧光显微镜下观察液滴亮度。
1.2.3.2 液滴分选 当液滴中的荧光强度具有一定区别时进行液滴分选。当液滴经过检测点时,在488 nm激光的照射下,液滴发射不同强度的荧光,荧光信号经PMT进行光电转换与信号放大作用后,电信号被采集卡处理,在软件上显示峰值。当液滴中荧光信号高于设定的阈值时,高压放大器输出高压电场,目标液滴流向“SORT”通道,非目标液滴流向“WST”通道,达到分选目的。后将分选出的液滴涂在LBHIS平板上,在30℃培养箱中倒置培养24 h。
1.2.4 分选菌株培养
1.2.4.1 孔板培养 将分选到的单菌落接种至CGX II(20 g/L葡萄糖)培养基的96浅孔板中,于30℃,800 r/min过夜培养,以初始OD600=0.2将种子液接种至含有1 mL的CGX II(60 g/L葡萄糖)的96深孔板中,于30℃,800 r/min培养24 h。
1.2.4.2 摇瓶培养 将复筛产量高的菌株接种到含有CGX II(20 g/L葡萄糖)培养基,于30℃,220 r/min过夜培养,以初始OD600=0.2将种子液接种至含有30 mL的CGX II(60 g/L葡萄糖)的250 mL摇瓶中,于30℃,220 r/min培养72 h,并分别取0、24、48和72 h样品1 mL测定L-Phe及OD600nm。
1.2.5 发酵参数测定 L-Phe生物传感器检测96孔板发酵液中L-Phe含量:配置缓冲溶液100 mmol/L Tris,pH 7.5,其中ATP 1 mmol/L,MgCl22.5 mmol/L,待测传感器0.5 μmol/L。将配置好的混合液转移至96孔荧光酶标板中,并加入10 μL去除菌体的发酵液上清,在荧光酶标仪中以激发光480 nm,发射光510 nm测试每个孔的荧光强度。高效液相色谱法(HPLC)检测孔板及摇瓶发酵液中L-Phe含量:(1)12 000 r/min离心2 min去除发酵液中的菌体,按照一定的倍数稀释发酵液。(2)用0.22 μm的过滤头对样品进行过滤,随后注入样品瓶中。(3)标准品的制备:精密称取L-Phe对照品0.165 2 g,定容于10 mL超纯水中,配置成浓度为100 mmol/L的L-Phe对照品储备液,并梯度稀释。(4)色谱条件:Waters C18(250 mm × 4.6 mm, 5 μm)色谱柱;流动相:乙腈∶水(10∶90),流速:0.5 mL/min;柱温:25℃;检测波长:192 nm,进样体积:20 μL,进样时间:15 min。
1.2.6 RBS强度测定 将pEC-XK99E系列质粒分别电转至谷氨酸棒杆菌ATCC 13032中,并涂布在LBHIS平板上(25 μg/mL卡那霉素),30℃培养箱中倒置培养24 h,至长出单克隆。将单克隆接种至LBHIS液体培养基(25 μg/mL卡那霉素)的96深孔板中,于30℃,800 r/min条件下培养16 h。随后,以同样接种量接种至含有1 mL的LBHIS液体培养基(25 μg/mL卡那霉素)的96深孔板中,于30℃,800 r/min条件下培养20 h。4 000 r/min离心15 min收集菌体,并用PBS洗涤3次,然后重悬于PBS缓冲液中。通过酶标仪(Synergy H4, Bio Tek)测量细菌悬浮液的OD600nm和GFP荧光信号(激发波长=488 nm,发射波长= 520 nm)。
2 结果
2.1 谷氨酸棒杆菌多基因诱导编辑底盘菌株的构建
前期实验室已将基因前端带有组成型启动子P11F和RBS为GGGGGGGG的aroFfbr和pheAfbr整合到了谷氨酸棒杆菌ATCC 13032的基因组中,整合位点如图2-A1所示,将其命名为C.g-0。将质粒pK18-ΔaroP::tktA-ppsA电转化至重组菌株C.g-0感受态中,整合位点如图2-A2所示,得到重组菌株C.g-1。随后将质粒pK18-ΔldhA::aroE-aroL-tyrB质粒电转化至C.g-1感受态中,整合位点如图2-A3所示,得到了重组菌株C.g-2。验证引物位置如图2所示,结果如图3所示。

图2 整合基因在基因组上位置及验证引物位置Fig.2 Positions of integrated genes on the genome and verification of primer positions

图3 C.g-1和C.g-2重组菌株两轮单交换验证Fig.3 PCR identification of the first-cross and second-cross of recombinant strain C.g-1 and C.g-2
2.2 RBS突变库编辑结果
对编辑混菌进行菌落PCR进行一代测序来检测每个基因的RBS的编辑情况。结果如图4所示,碱基编辑器对每个基因前的RBS都进行了编辑,产生了富含G/A的RBS序列。由于蛋白质翻译起始阶段依赖于序列的核糖体募集,所以RBS可以直接调控基因的表达,碱基编辑器可以同时对多个基因进行靶向编辑,产生大量具有不同表达水平的目标基因的组合,从而大规模地扰动不同代谢模块。

图4 一代测序基因的RBS编辑结果Fig.4 Sequencing result of different RBSs of genes
2.3 RBS突变文库的液滴生成、培养观察及分选
液滴的生成过程如图5-A所示,将油、传感器、菌液分为3通道进行共包埋。结果如图5-B所示,获得单分散液滴具有良好的均一性,其中有菌的液滴比例大约为20%,并且只有极少数两个及以上的菌被封装在同一个液滴中,基本符合泊松分布的分布规律。RBS突变文库在0 h时,液滴荧光基本一致,所有液滴的荧光亮度都较低;在6 h时,RBS突变文库中有菌的液滴与空液滴相比有一定亮度,但由于生长时间较短,液滴中的菌未能充盈整个液滴;在12 h时,观察到菌体数目变多,基本可充盈整个液滴;在16 h时,RBS突变文库中部分液滴荧光亮度较12 h更明显;在24 h时,所有液滴的荧光骤减,这可能是由于传感器荧光淬灭造成的[23-24]。因此,选择16 h进行后续的液滴分选(图5-C),此时突变库的液滴荧光强度分布和分选情况如图5-D所示。将荧光强度最高的前5%设置为分选阈值,从约7万个液滴中分选得到大约3 000个液滴(收集于一个EP管中),经过LBHIS琼脂板的进一步培养,获得大约100个单克隆。

图5 液滴收集、培养与观察及液滴分选Fig.5 Droplet collection, culture and observation, and droplet sorting
2.4 L-Phe高产菌株获得及验证
将分选到的菌株单克隆接种到96孔板中发酵培养24 h,首先利用L-Phe生物传感器进行复筛,并选取荧光值排序最高的两株菌株以及荧光值居中的两株菌,使用HPLC测定L-Phe产量,结果如图6-A所示,不同菌株的HPLC测定的L-Phe产量与生物传感器得到的荧光值呈正相关性。相较于对照菌株C.g-2,所有突变菌株在L-Phe标品的保留时间(~9.7 min)处的吸收峰均有明显的提升(图6-B)。通过标准曲线的拟合换算,突变株C.g-Mut1、C.g-Mut2、C.g-Mut3、C.g-Mut4的孔板发酵L-Phe产量分别为0.14、0.35、1.05和1.58 mmol/L,相比对照菌株C.g-2(0.008 mmol/L),提高了16.50、42.75、130.25和196.50倍。为了评估突变菌株的发酵性能,对其摇瓶发酵,各突变株的生物量在48 h后趋于稳定(图6-C)。产量如图6-D所示,24 h和48 h摇瓶发酵与孔板发酵成正相关,72 h C.g-Mut1产量高于C.g-Mut2;72 h时C.g-Mut1、C.g-Mut2、C.g-Mut3、C.g-Mut4的摇瓶发酵L-Phe产量分别为2.06、1.30、4.42和7.44 mmol/L(1.23 g/L)。相比对照菌株C.g-2(0.6 mmol/L),提高了2.43、1.17、6.37和11.40倍。各个突变菌株中每个基因的RBS的测序结果如表3所示,任何一株筛选得到的突变株,均有至少5个基因的RBS发生编辑,这与2.3节针对混菌的测序结果基本一致。所有突变株的tktA、pheAfbr、aroFfbr基因的RBS均发生了一定程度的编辑。

表3 突变菌株RBS测序Table 3 RBS sequencing for mutated strains
2.5 RBS强度分析
对起始菌株C.g-2和筛选的L-Phe高产菌株C.g-Mut4进行RBS强度测试,使用RBS强度报告系统表征RBS强度。结果如图7所示,与原始C.g-2的GGGGGGGG相比,突变菌株C.g-Mut4的aroE、aroL、tktA翻译效率分别提高了58%、34%、47%,tyrB、ppsA、pheAfbr、aroFfbr翻译效率分别提高了4.37、8.62、6.24、22.29倍。L-Phe产量的提升与所有关键基因翻译效率的提高呈正相关。其中,突变后的aroFfbr基因的RBS的单位细胞荧光信号的绝对值最高,翻译效率提升幅度最大,pheAfbr和aroFfbr的表达强度远远高于另外5个关键基因,说明pheAfbr和aroFfbr是L-Phe生物合成途径中至关重要的两个基因。

图7 使用染色体GFP报告基因测定C.g-2和C.g-Mut4的RBS的强度Fig.7 RBS intensity for C.g-2 and C.g-Mut4 determined using chromosomal GFP reporter genes
3 讨论
3.1 多位点同步编辑对于平衡不同代谢模块的优势讨论
本研究将大肠杆菌MG1655来源的解除了反馈抑制的pheAfbr和aroFfbr基因,以及L-Phe合成途径中的其他5个关键基因整合到了谷氨酸棒杆菌13032的基因组中,并通过碱基编辑技术编辑每个基因前端的RBS,从而协同调控7种酶在谷氨酸棒杆菌中的表达,并通过液滴微流控结合L-Phe生物传感器对RBS突变文库进行筛选,从而得到L-Phe高产菌株。在L-Phe生物合成的研究中,莽草酸途径的DAHPS是公认的限速酶,一方面受到L-Phe对酶活性的反馈抑制,另一方面其表达水平也受到严格调控。而随着DAHPS的解调,中心代谢途径中代谢物丰度相对较低的前体物E4P的供给可能不足,因此有大量研究尝试对tktA基因过表达。随着莽草酸途径通量的增加,分支酸途径中同样受到L-Phe对酶活性的反馈抑制的CM/PDT又将成为新的限速步骤[25]。本研究结果中所有L-Phe产量提升的突变株的pheAfbr、aroFfbr、tktA的RBS均发生了编辑,表明通过协同调控中心代谢途径(供给E4P)、莽草酸途径(供给分支酸)和分支酸途径(合成L-Phe)对于高产L-Phe具有重要的作用,这与前人的研究一致[26]。此外,本研究还从这3个模块中选取对L-Phe合成有利的PEP合酶编码基因ppsA(PEP回补途径),莽草酸脱氢酶编码基因aroE、莽草酸激酶编码基因aroL、苯丙氨酸氨基转移酶编码基因tyrB,一同进行协同调控与筛选。从不同菌株7个基因的RBS分布上来看,不同RBS的组合对L-Phe产量的影响很大,而产量最高的突变株C.g-Mut4的7个基因的RBS均发生了编辑,这暗示了越多基因的协同调控将越有利于不同模块之间通量达到平衡,也充分说明了BETTER技术在多基因精细调控方面的优势。此外,C.g-Mut2和C.g-Mut3的染色体上ΔaroP位点的人工簇中缺失了ppsA基因,这可能是由于BETTER技术进行碱基编辑时nCas9会造成多点单链断裂[15],当ΔaroP位点的ppsA基因的上下游存在重复出现的启动子P11F和终止子TrrnB时,有概率会发生同源重组将ppsA基因弹出基因组,暗示着在某些多基因协同表达的组合中,ppsA基因的强表达可能反而不利于L-Phe的积累,相关机理有待进一步研究。
3.2 突变株C.g-Mut4高产L-Phe的机制分析
RBS强度分析结果表明,来源于大肠杆菌MG1655的解除反馈抑制的aroFfbr和pheAfbr基因,突变后的RBS单位细胞荧光信号绝对值最高,说明DAHPS和CM/PDT的反馈抑制的解除与强表达对于L-Phe的高产是至关重要的,这与现在研究的普遍认识是相符的[25]。此外,PEP合酶编码基因ppsA和苯丙氨酸氨基转移酶编码基因tyrB的突变后的RBS的单位细胞荧光信号,相比于对照菌株的RBS,分别提高了8.62和4.37倍。其相关机理可能是随着DAHPS的解调,中心代谢途径中代谢物丰度相对较低的前体物PEP的供给可能不足,因此会筛选出ppsA表达水平提高的突变;而随着CM/PDT的解调,L-Phe合成的直接前体物苯甲酮酸的积累量提高,需要上调tyrB的表达水平,因此筛选到了相关突变。转酮酶编码基因tktA、莽草酸脱氢酶编码基因aroE与莽草酸激酶编码基因aroL的突变后RBS的单位细胞荧光信号,虽然相比于对照也均提升了约50%,但仍处于较低水平。这说明在C.g-Mut4菌株中,这些基因暂时不是L-Phe合成的限速步骤。
3.3 应用BETTER技术构建高产L-Phe的可行性分析
相比于目前主流的基于游离型质粒或者多轮迭代的基因组编辑对关键基因强化表达方式,本研究中采用的碱基编辑介导的多基因调控技术与液滴微流控高通量筛选技术,可以在很短的周期内,获得稳定、安全、高效的高产菌株,而且后续发酵不需要额外添加抗生素和诱导剂。产量最高的突变株C.g-Mut4,在孔板评价阶段,与对照菌株C.g-2相比提高196.50倍。在后续的摇瓶发酵评价中,虽然没有经过发酵工艺优化(采用最通用的基本盐培养基CGXII),也可以积累7.44 mmol/L(1.23 g/L)的L-Phe,较对照菌株C.g-2产量提高11.40倍,这充分证明了利用碱基编辑器同时调节多基因的表达水平并筛选组合文库是提高谷氨酸棒杆菌产L-Phe性能的一种行之有效的策略。在未来的研究工作中可将C.g-Mut4作为底盘菌株,在此基础之上继续进行代谢工程改造,例如提高L-Phe转运系统的效率、降低L-Phe竞争性物质的积累等,结合发酵工艺优化与放大,有望获得更具工业生成潜力的高产菌株。
4 结论
本研究通过同源重组技术,将L-Phe生物合成途径中的7个关键基因和相关调控元件分为3个人工模块,分别整合到谷氨酸棒杆菌ATCC 13032基因组的不同位点中,使用碱基编辑技术靶向编辑7个基因前端的RBS从而同时实现3个代谢模块的模块内与模块间的协同调控,并结合液滴微流控技术对所得突变文库进行高通量筛选,成功得到一株产量为7.44 mmol/L(1.23 g/L)的L-Phe的高产菌株,较对照菌株C.g-2产量提高11.40倍。
