嵌合抗原受体-T细胞免疫疗法在多发性骨髓瘤中的研究进展
2023-10-20安霖综述张宏伟审校
安霖 综述,张宏伟 审校
1.山西医科大学第二临床医学院,山西太原 030001;2.山西医科大学附属肿瘤医院血液科,山西太原030013
多发性骨髓瘤(multiple myeloma,MM)是浆细胞恶性增殖性疾病,是常见的血液系统恶性肿瘤[1],其特征为骨髓中克隆性浆细胞异常增生,绝大部分病例存在单克隆免疫球蛋白或其片段(M 蛋白)分泌,导致相关器官或组织损伤,常见临床表现为骨痛、贫血、肾功能损害、血钙增高和感染等。近年来,MM患者普遍接受化疗、自体干细胞移植、蛋白酶抑制剂、免疫调节药物和单克隆抗体治疗,但大多数患者最终会复发[2]。
免疫疗法已经发展为一种实用的癌症治疗方法[3-4]。嵌合抗原受体(chimeric antigen receptor,CAR)-T 细胞免疫疗法是一种快速出现且极具前景的免疫治疗方法,在B 细胞恶性肿瘤中显示出前所未有的效果[5-8],延长了患者的生存期和缓解期,因此激发了包括MM 在内的其他癌症上CAR-T 细胞免疫疗法的研究。CAR 是通过基因工程技术获得的嵌合蛋白,主要由3 部分组成:细胞外抗原识别结构域[通常为单克隆抗体的单链可变片段(single-chain variable fragment,scFv)、铰链区]和CD8 的跨膜结构域以及细胞内信号转导结构域;CAR-T 细胞免疫疗法是一种使用转基因效应T 细胞治疗肿瘤的新策略,识别并杀死表达特定抗原的癌细胞,且不受主要组织相容性复合物(major histocompatibility complex,MHC)限制。
本文对CAR-T 细胞免疫疗法在MM 中的研究进展、治疗MM的局限性及其优化作一综述。
1 CAR-T不同靶点在MM中的应用
1.1信号淋巴细胞活化分子受体家族7(signalling lymphocyte activation molecule family 7,SLAMF7)/白细胞分化抗原2亚集1(cluster of differentiation 2 subset 1,CS1)/白细胞分化抗原319(cluster of differentiation 319,CD319) CS1/SLAMF7/CD319蛋白在造血干细胞上不表达,表达仅限于造血系统,包括成熟的NK 细胞、树突细胞、浆细胞和一些T 细胞[9-10],由于在MM中过度表达,因此是CAR-T 细胞免疫疗法治疗的合理靶点。Elotuzumab是一种人源化CS1抗体(克隆体HuLuc63),在对复发/难治性MM(relapsed or refractory multiple myeloma,RRMM)患者进行的1b-2 期研究中,实验组采用Elotuzumab、来那度胺和地塞米松联合治疗,随机分配321 名患者;对照组单用来那度胺和地塞米松治疗,随机分配325 名患者。在中位数为24.5 个月的随访后,实验组1 年无进展生存率为68%,而对照组为57%;2 年后,这2 个比率分别为41%和27%。实验组的中位无进展生存期为19.4个月,而对照组为14.9 个月,实验组相对于对照组进展或死亡的风险比(hazard ratio,HR)为0.70(95%CI:0.57 ~0.85,P<0.001)。实验组总有效率为79%,而对照组为66%(P<0.001)。2 组中常见的3 或4级不良事件为淋巴细胞减少、中性粒细胞减少、乏力和肺炎。实验组有33 名患者(10%)出现输注反应,29名患者出现1或2级反应。接受实验组治疗RRMM患者的疾病进展或死亡风险显著相对降低30%,并且无严重相关毒性[11-12],表明用CAR-T细胞靶向CS1是一种安全、有效的抗骨髓瘤疗法。
1.2B细胞成熟抗原(B cell maturation antigen,BCMA)BCMA 是一种Ⅲ型跨膜蛋白,属于肿瘤坏死因子受体(tumor necrosis factor receptor,TNFR)家族,主要在终末分化的正常B 细胞和浆细胞上表达,同时也是MM 细胞上高度表达的表面抗原[13-15]。美国国家癌症研究所BRUDON 等[16]进行了抗BCMA-CAR-T 细胞的第1次人体试验,总缓解率(overall response rate,ORR)为81%,63%表现出非常好的部分缓解(very good partial remission,VGPR)或完全缓解(complete remission,CR),中位无进展生存时间(progression-free survival,PFS)为7.1个月(NCT02215967)。目前主要的BCMA CAR-T细胞临床试验见表1[17-22]。
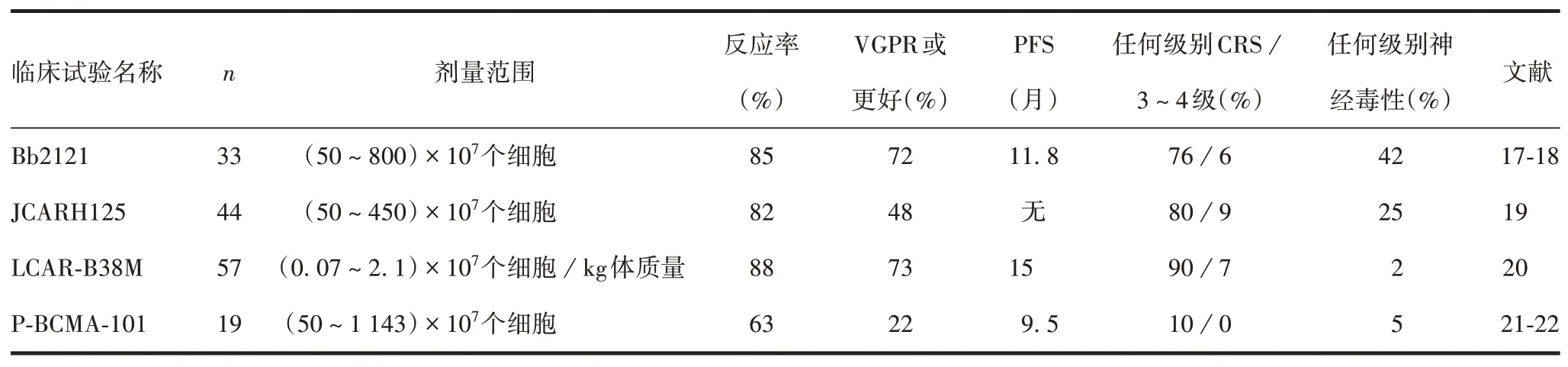
表1 BCMA CAR-T细胞临床试验背景资料Tab.1 Background information of clinical trial of BCMA CAR-T cells
1.3白细胞分化抗原19(cluster of differentiation 19,CD19) CD19(也称B4)是一种B细胞特异性跨膜蛋白,属于免疫球蛋白超家族。CD19 是一种增强B 细胞受体反应的共刺激分子[23],在B 细胞发育的几乎所有阶段均有表达,仅在一小部分骨髓瘤细胞上表达。研究表明,抗CD19 CAR-T(anti-CD19 CAR-T cell,CTL019)可在体外消除CD19 阳性骨髓瘤细胞[24]。一项研究中纳入的所有受试者均患有RRMM,且之前接受过自体造血干细胞移植(autologous stem cell transplant,ASCT)患者的PFS 不到1 年。受试者在补救性高剂量美法仑和自体干细胞移植后接受CTL019治疗,结果显示,CTL019 可通过靶向和促进针对骨髓瘤增殖细胞的2 次免疫应答来延长对MM 治疗的应答持续时间[25]。
1.4G 蛋白偶联受体家族C 组5 成员D(G proteincoupled receptor class C group 5 member D,GPRC5D)GPRC5D 属于一种孤儿受体,暂未发现其内源性配体,为7 次跨膜蛋白,在正常组织的表达仅限于毛囊区域[26]。2019 年,纪念斯隆凯特琳癌症中心(Memorial Sloan Kettering Cancer Center,MSKCC)和瑞典癌症研究所(Swedish Cancer Institute)的科学家公布了一项GPRC5D CAR-T 细胞免疫疗法的验证性研究成果,显示GPRC5D 在MM 上表达。其结果还进一步表明,在BCMA 抗原丢失导致的肿瘤复发模型中,GPRC5D 靶向的CAR-T 细胞免疫疗法可抑制肿瘤逃逸,证实了GPRC5D 和BCMA 独立表达的特点[27-28]。目前针对CD3 和GPRC5D 的双特异性抗体的剂量递增研究正在进行中(NCT03399799)。
1.5白细胞分化抗原138(cluster of differentiation 138,CD138) CD138是一种细胞表面表达的硫酸乙酰肝素蛋白聚糖,属于黏结蛋白聚糖(Syndecan)家族,在前体B细胞和浆细胞上表达[29]。一项针对anti-CD138 CAR-T 细胞免疫疗法治疗的5 名RRMM 患者的临床试验结果显示,4名患者病情稳定(NCT01886976)[30];另一项使用anti-CD138 CAR-T 细胞免疫疗法治疗RRMM患者的临床试验正在进行中(NCT036723-18)[31]。
1.6白细胞分化抗原38(cluster off differentiation 38,CD38) CD38的组织细胞分布很有特点,在浆细胞、T 细胞、NK 细胞、树突细胞等中表达量较高,研究表明,这些免疫细胞中的CD38对于T细胞、树突细胞和中性粒细胞的迁移均不可或缺[32]。在非实体瘤中,如慢性淋巴细胞白血病(chronic lymphocytic leukemia,CLL)、套细胞淋巴瘤、急性髓系白血病、急性淋系白血病等肿瘤细胞中均有较高的表达,但CD38表达最高的为MM,且与MM 预后负相关,这使得CD38成为MM 靶向治疗的良好靶点。目前正在招募一项针对RRMM 患者的抗CD38 CAR-T 细胞免疫疗法的开放性Ⅰ期单剂量递增安全性研究(NCT0346-4916)[33]。
1.7κ 轻链 B 淋巴细胞和大多数低度恶性淋巴瘤细胞表达带有κ 或λ 轻链的单克隆免疫球蛋白。研究表明,抗κ 轻链CAR-T 细胞在体外和体内对免疫球蛋白肿瘤细胞系均具有细胞毒活性[34]。在一项临床试验中,使用抗κ轻链CAR-T 细胞免疫疗法治疗7例MM患者,4名患者病情稳定,持续2 ~17个月(NCT-00881920)[30]。
综上所述,与其他针对MM 和RRMM 的CAR-T细胞免疫疗法相比,抗BCMA CAR-T 细胞免疫疗法具有一定优势:①BCMA 在终末分化的B 细胞上表达,因此抗BCMA CAR-T 细胞免疫疗法在临床试验中具有更好的抗肿瘤功效;②BCMA 的表达几乎仅限于B 细胞,且在恶性浆细胞上的表达高于正常细胞[35-36],有利于降低抗BCMA CAR-T 细胞免疫疗法的靶向脱瘤毒性,从而增加安全性;③用于治疗MM和RRMM 的抗BCMA CAR-T 产品较多,临床经验也相对较多。但抗BCMA CAR-T 细胞免疫疗法仍存在局限性。
2 CAR-T治疗MM引起的局限性
CAR-T 细胞免疫疗法是治疗MM 的新型疗法,尽管取得一定效果,但CAR-T细胞免疫疗法的改进仍然存在重大挑战和机遇。虽然大多数患者对治疗有反应,但最终会发展为复发或难治性疾病。毒性也仍然是CAR-T 细胞免疫疗法的主要限制。抗原逃逸、CRS 和免疫效应细胞相关神经毒性综合征(immune effector cell associated neurotoxicity syndrome,ICANS)通常需要住院治疗,加重了经济负担。
2.1抗原逃逸 CAR-T 细胞免疫疗法最具挑战性的局限性之一是肿瘤对单抗原靶向CAR构建体的耐药性的发展。尽管最初单抗原靶向的CAR-T细胞免疫疗法可提供高应答率,但用其治疗的患者中,大部分的恶性细胞显示出靶抗原表达的部分或完全丧失,这种现象被称为抗原逃逸。在接受BCMA靶向CART 细胞免疫疗法治疗的MM 患者中,也观察到BCMA表达下调或缺失。
2.2CAR-T细胞免疫疗法治疗的相关毒性 CRS是最常见的严重急性CAR-T 细胞相关毒性。CRS 是由活化的T 细胞或肿瘤细胞本身大量释放IFNγ 触发的[37]。分泌的IFNγ 诱导其他免疫细胞的激活,最重要的是巨噬细胞[8]。活化的巨噬细胞会产生过量的额外细胞因子,如IL-6、TNF-α 和IL-10,从而导致发热、疲劳、血管渗漏、心肌病、低血压和凝血障碍等临床症状[38-40]。
神经毒性现被称为ICANS,是CAR-T 细胞输注后第二常见的不良事件[41],目前病理生理学机制尚不完全清楚,但更多的数据为细胞因子介导的内皮激活提供了强有力的证据,这些激活会导致凝血障碍、毛细血管渗漏和血脑屏障(blood brain barrier,BBB)破坏,从而导致高浓度的全身性细胞因子进入脑脊液[42]。其临床症状多种多样,最常见的症状包括脑病、头痛、谵妄、焦虑、震颤、失语,在CAR-T细胞免疫疗法的临床试验中也观察到了其他神经毒性表现,如意识水平下降、困惑、癫痫发作和脑水肿[43]。
脱靶毒性包括在大多数患者中发生的血细胞减少,并且在患者中还报告了主要出血的并发症,包括胃肠道出血,颅内出血和血胸[44-45]。BCMA 几乎在成熟的B 淋巴细胞、浆细胞和MM 细胞上独家表达,这可能导致继发性低丙种球蛋白血症,因为健康的浆细胞也可能受到BCMA 指导的CAR-T 细胞的影响,而另一个潜在的非靶点并发症是插入性肿瘤发生的可能性[46-47]。研究表明,在造血干细胞基因治疗X 连锁严重联合免疫缺陷和慢性肉芽肿疾病的背景下,已确定人类细胞中插入肿瘤发生的风险[48-52]。在大多数情况下,与逆转录病毒载体插入原癌基因LMO-2(LIM domain only 2)附近有关[52],将转基因插入分化的T 细胞也有诱发恶性转化的风险。但至今仍未见报道在输注转基因T 细胞后发生转化的案例。值得注意的是,LMO-2致癌基因在T 细胞中沉默,使得该位点不太可能成为逆转录病毒整合的位点。在实践中,使用转基因T 细胞具有长达10 年的安全性,但并无证据表明载体诱导的永生化、克隆扩增或与生长控制或转化有关的基因附近的整合位点富集。综上,插入的风险基因转移至T 细胞后的肿瘤发生率似乎很低,但研究人员必须遵守当前临床试验设计中规定的严格监测流程,与此同时需要对接受CAR-T 细胞免疫疗法治疗的患者进行长期监测[53]。
2.3免疫抑制微环境 在肿瘤微环境中,许多驱动免疫抑制的细胞类型可浸润实体瘤,包括髓源性抑制细胞(myeloid-derived suppressor cells,MDSCs)、肿瘤相关巨噬细胞(tumor-associated macrophages,TAMs)和调节性T 细胞(T-regulatory cells,TREGs),这些驱动免疫抑制细胞浸润肿瘤细胞从而使肿瘤促进细胞因子、趋化因子和生长因子的产生[54]。此外,免疫检查点途径如程序性死亡受体-1(programmed cell death-1,PD-1)或细胞毒性T 淋巴细胞相关蛋白-4(cytotoxic T-lymphocyte associated protein-4,CTLA-4)可降低抗肿瘤免疫。对CAR-T细胞免疫疗法治疗无反应或反应弱的主要原因之一是T 细胞增殖不良或T 细胞在患者体内的持久性。因此,结合CAR-T 细胞和检查点阻断的免疫治疗被认为是下一个免疫治疗的前沿,因为其提供了强大的免疫反应所需的2 个元素:CAR-T 细胞,提供浸润;PD-1/程序性死亡受体-配体1(programmed cell death-ligand 1,PD-L1)阻断,确保T细胞的持续存在及功能[55]。
3 CAR-T细胞治疗MM的优化
3.1提高CAR 的设计 CAR-T 细胞的制备是通过基因工程技术在淋巴T 细胞中加入1 个能识别肿瘤细胞、同时激活T 细胞、最终杀死肿瘤细胞的嵌合受体。相关基因工程技术包括慢病毒、γ 逆转录病毒、mRNA 转染、mRNA 电转等。随后进行免疫表型分析,以确保成功赋予T 细胞CAR 和溶细胞活性,制备完成后通过细胞培养技术在体外大规模培养CAR-T细胞,使其数量达到治疗水平[56]。第1 代CAR-T 细胞仅包含CD3ζ 信号结构域,缺乏增殖特征[57]。当前和常规生产的CAR-T 产品整合了1(第2 代)或2个(第3 代)共刺激结构域(4-1BB、CD28 或OX-40),以促进有效的CAR-T 细胞信号传导以及其持久性和功效。第4 代CAR 还表达可促进T 细胞增殖的细胞因子,如IL-2、IL-12,第5 代CAR 的设计旨在同时激活T 细胞表面受体(T cell receptor,TCR)、共激域CD28 和细胞因子三重信号,因为这是T 细胞结合抗原时被激活的前提条件。研究人员证实,在血液肿瘤和实体瘤模型中,与第2 代CAR 相比,第5 代CAR通过基因改造增强了T 细胞增殖、存活以及抗肿瘤效果[58-61]。
3.2替代和双靶向CAR-T 细胞 双特异性CAR 疗法、复合CAR 疗法、CAR 联合疗法以及其他抗BCMA CAR-T产品,如Descartes-08、ALLO-715等,均进入临床试验阶段。但这些疗法可能会增加靶向外肿瘤毒性,因此使用这些疗法的最重要标准是患者自身的状况[62]。
3.3联合治疗 许多临床研究正在评估已建立的CAR-T 细胞免疫疗法与一些潜在的添加剂或增效剂的组合,这些添加剂或增效剂被认为可增强CA 细胞的抗肿瘤活性,减少T 细胞衰竭或减轻毒性。研究CAR-T 产品的相关临床试验正在进行中,包括检查点抑制剂、免疫调节剂和Bruton 酪氨酸激酶(Bruton's tyrosine kinase,BTK)抑制剂。其他旨在降低CRS 和神经毒性风险或严重程度的治疗也在进行中,如抗IL-1R 药物Anakina 正在进行积极的临床研究[56]。
通过PD-1和PD-L1 抑制剂破坏PD-1/PD-L1免疫检查点已在包括霍奇金淋巴瘤在内的许多恶性肿瘤中显示出活性。一些临床试验正在研究检查点抑制剂与CAR-T 细胞的联合治疗,以减少CAR-T 细胞耗竭并促进更强大的抗肿瘤反应,初步结果显示具有足够的安全性,但仍需大规模的研究来确定该组合是否会产生更好的效果[56,63]。
BTK 抑制剂Ibrutinib 联合CAR-T 治疗已在临床前模型中被证明可减少与CAR-T相关的细胞因子的产生。最近将Ibrutinib 与CAR-T 细胞免疫疗法一起用于Ibrutinib-难治性CLL 的Ⅰ期研究,与另一组未接受Ibrutinib 的队列相比,显示出相似的响应率和较低的CRS 严重程度,并且具有更好的CAR-T 增殖趋势[63],有关CLL 和NHL 的更大的前瞻性临床试验正在进行中,以进一步评估这些初步发现[56,64]。
临床前数据还表明,来那度胺等免疫调节剂也可增强CAR 细胞的抗肿瘤作用,目前进行的研究正在评估这种免疫调节剂与其他免疫调节剂以及已建立的CAR结构的安全性和有效性[6]。
4 小结与展望
随着CAR-T 细胞免疫疗法的发展,近年来该疗法在治疗MM 时,与其他化疗相比取得了明显进步。至今为止,包括双特异性抗体在内的众多免疫疗法,如双特异性T 细胞Enger,使用树突状细胞疫苗、自体CAR-T 细胞、同种异体CAR-NK 细胞与检查点抑制剂的双体过继细胞疗法,已被开发用于治疗MM,并且目前正在进行或预计将进行各种临床试验。未来将根据联合疗法以及同种异体CAR-T 或NK 细胞疗法的疗效,有希望改变MM 的治疗模式,以提高患者生存率、改善患者生活质量。
