计算机辅助药物设计技术在雄激素受体动态机制探讨和药物发现中的研究进展
2023-10-20富炜涛柯迪李丹侯廷军
富炜涛,柯迪,李丹 ,侯廷军
(1.江苏威凯尔医药科技有限公司创新药研发中心,江苏 南京 211800;2.浙江大学药学院,浙江 杭州 310058;3.浙江大学金华研究院,浙江 金华 321000)
雄激素受体(androgen receptor,AR)及其配体雄激素在男性第二性征、肌肉和骨骼等发育过程中具有重要的作用[1-2]。异常的AR 信号与多种疾病密切相关,如前列腺癌、雄激素不敏感综合征(androgen insensitivity syndrome,AIS)、脊髓延髓肌萎缩症等[3-5]。其中,前列腺癌位列男性肿瘤发病率第2 名,且是男性致死率第5 名的恶性肿瘤[6]。在我国,前列腺癌的发病率和死亡率呈现逐年递增的趋势[7]。作为威胁人类生命健康的重大疾病,前列腺癌的新型治疗策略是各大制药公司和学术机构研究的热点。其中,AR 信号通路在前列腺癌的发生发展中发挥关键的调控作用,靶向AR 的小分子药物开发已获得了巨大的成功,并且已有多款上市药物在全球范围内使用,包括第1 代的羟基氟他胺(1)、尼鲁米特(2)和R-比卡鲁胺(3),以及第2 代的恩杂鲁胺(4)、阿帕鲁胺(5)和达洛鲁胺(6)(见图1)[3]。
图1 第1 代和第2 代靶向雄激素受体上市药物化学结构式Figure 1 Structures of the first and second generations of approved androgen receptor antagonists
AR 属于核受体家族,由920 个氨基酸残基组成,分为3 个主要结构域:N-端结构域(N-terminal domain,NTD)、DNA 结合结构域(DNA-binding domain,DBD)和配体结合域(ligand-binding domain,LBD)。AR 信号通路中,AR 在非活性状态下与热休克蛋白结合,而当有雄激素与AR 结合时,AR 会从热休克蛋白解离,并募集共激活剂,以AR 二聚体在核内与雄激素响应元件相结合,启动相关转录程序,发挥生物学功能[3]。
AR 的DBD 和LBD 的6 个结合位点已经有多种新型小分子拮抗剂通过计算机辅助药物设计(computer aided drug design,CADD)相关技术被发现(见图2),这些位点包括AR-DBD 结合区域P-Box 和D-Box 附近的2 个位点,AR-LBD 的配体结合口袋(ligand-binding pocket,LBP,已上市小分子药物的结合位点)、激活功能区2(activation function 2,AF2)、结合功能区3(binding function 3,BF3)和二聚体界面结合口袋(dimer interface pocket,DIP)[3]。
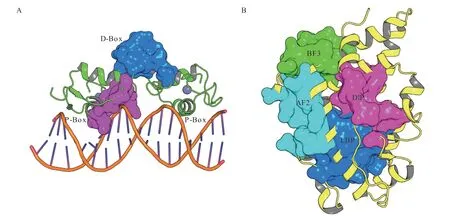
图2 雄激素受体的DNA 结合结构域和配体结合域已发现小分子拮抗剂的6 个结合位点Figure 2 The 6 binding pockets discovered for the binding of small-molecule antagonists located at AR-DBD and AR-LBD
无论是第1 代还是第2 代上市药物,在用药一段时间后,AR基因的扩增、药物分子结合位点的氨基酸残基点突变、剪接变异体的出现等,导致这些AR 靶向药物发生不同程度的耐药,甚至从拮抗剂转变为激动剂,极大限制了临床治疗效益[8]。为了提供新的治疗策略,多种多样的技术应用于靶向不同新位点的新型拮抗剂的开发[9-10]。其中,CADD技术在雄激素受体-配体动态机制的探讨,特别是在耐药机制方面的研究,以及新型拮抗剂的发现和新位点探索上展现出独特的优势。综述CADD 技术在AR 相关的机制探讨和新位点新活性拮抗剂发现方面的应用,为后续CADD 技术在AR 药物开发中的应用以及更多核受体中的应用提供重要参考。
1 常用的计算机辅助药物设计技术
1.1 结构预测和构建
当靶标受体的结构是未知且仅有序列信息的情况下,通过同源建模和人工智能(artificial intelligence,AI)预测是目前常用的有效方法。同源建模是以实验获得的同源受体三维结构信息为模板,构建目标序列三维结构的方法[11]。AI 结构预测是通过AI 技术对已经解析的受体三维结构和序列之间的关系进行训练推理,用于预测未解析的三维结构。AI 结构预测的蛋白质结构已经达到原子水平的准确度,代表性的技术如AlphaFold,RoseTTAFold,ColabFold,Uni-Fold 等[12-15]。
1.2 分子对接
分子对接是依据结构形状匹配和能量匹配,预测受体和配体之间结合模式的一种技术。在个人电脑上,分子对接即可快速预测受体分子和配体分子之间的结合模式。配体可以是化学小分子、多肽、蛋白、DNA 和RNA。在蛋白-小分子预测方面,经典的基于搜索的对接方法有AutoDock,Vina,Glide 等,以及新型的基于扩散生成模型DiffDock 等[16-17]。
1.3 基于结构的虚拟筛选
基于结构的虚拟筛选是指在有合适受体晶体结构或者构建的合理模型结构的基础上,运用分子对接技术,将化合物库(含数万到数亿)的分子一一对接到受体,并按照打分函数进行打分排序,优选排序靠前、结构新颖的化合物进行实验验证。这一技术能够快速筛选大规模的化合物库,优选合适的潜在活性化合物,在效率和成本上相比基于实验的高通量筛选具有明显的优势[17-18]。
1.4 基于配体的虚拟筛选
基于配体的虚拟筛选依据作用于特定靶标的已知活性分子构建合适的活性预测模型,以筛选出结构多样且具有生物活性的分子。随着AI 技术的不断发展,多种多样活性预测模型和分子生成等技术的不断涌现,基于配体的虚拟筛选在定量构效关系分析、药效团模型构建等传统模型基础上,应用范围得到了极大拓展[19-21]。
1.5 分子动力学模拟
基于同源建模或者AlphaFold 等AI 技术构建的结构或者晶体结构通常是静态结构,难以反映研究对象的动态过程,而分子动力学(molecular dynamics,MD)模拟是研究这样动态过程的重要技术。MD 模拟基于分子力场,通过在模拟过程中不断求解牛顿运动方程,以模拟研究对象的动态过程。对动态过程的研究,能够更清晰地了解研究对象的动态机制,如受体氨基酸残基点突变对配体结合的耐药机制、受体-配体结合解离的机制等[22]。此外,MD 模拟还能辅助结构相对单一的受体晶体结构或者构建的模型结构获得多样性的动态结构。
1.6 结合自由能预测
考察受体与配体结合强度的关键表征指标即两者的结合自由能。结合自由能常用的方法有打分函数(传统的和新型基于机器学习的打分函数)、基于过程的能量计算(伞形采样、拉伸分子动力学模拟等)和两点式自由能计算[分子力学/广义波恩表面积(molecular mechanics/generalized Born surface area,MM/GBSA)、分子力学/泊松-波尔兹曼表面积(molecular mechanics/Poisson Boltzmann surface area,MM/PBSA)、炼金术自由能(热力学积分、自由能微扰)][23-26]。不同的自由能预测方法有不同的适用范围,如打分函数预测速度快,常用于大规模虚拟筛选;炼金术自由能由于计算速度慢且对计算分子间结构的相似性要求高,同时精度较高,因此,常用于苗头分子的理性设计;而能够较好平衡计算精度和效率的MM/GBSA 和MM/PBSA方法在基于结构的虚拟筛选重打分和基于结构的理性设计中被广泛应用[27]。
2 计算机辅助药物设计技术在雄激素受体靶标机制探索方面的应用
由于AR-LBD 的晶体结构解析相对完整以及AR-LBD 的重要作用,CADD 技术在基于AR 靶标机制的研究方面主要是围绕AR-LBD 展开。ARLBD 二聚体由2 个同源AR-LBD 组成,每个单体具有3层折叠结构,包括由螺旋H1和H3形成的第1层,由螺旋H4,H5,H8 和H9 形成的中间层,以及由螺旋H6,H7,H10-11 和H12 形成的第3 层(见图3A)。
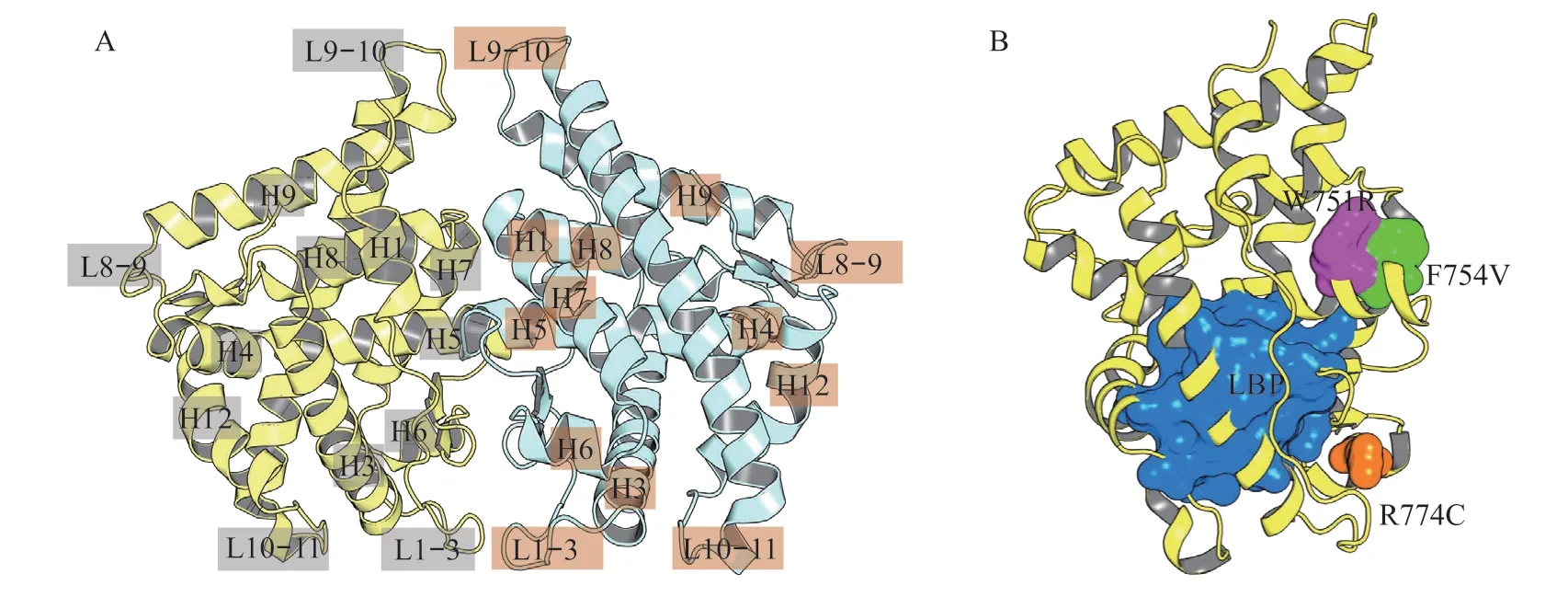
图3 AR-LBD 二聚体结构以及雄激素不敏感综合征相关点突变位置Figure 3 The structure of AR-LBD dimer and the location of androgen insensitivity syndrome-related mutation
2.1 雄激素不敏感综合征机制的研究
AIS 属于罕见的46XY 性发育异常疾病,主要表现为染色体46XY,但表现女性的第二性征。依据AIS不同的严重程度,分为轻型、部分型和完全型。AR作为关键基因,其无义突变、错义突变、剪接变异体、基因缺失、基因插入与AIS 密切相关[28]。仅在AR-LBD,就存在40 余个与AIS 相关的点突变[29]。作为实验验证的重要补充,MD 模拟在机制阐明方面发挥了重要作用,有助于探究这些突变如何影响AR 的结构或者雄激素的结合。
Arg774 突变为Cys774(R774C)是导致AIS 的重要点突变,而该突变并不在AR 的LBP 位点,即不在雄激素结合位点,而R774C 如何影响AR 的功能,尚无法解释(见图3B)。为此,Wu 等[30]设置不同的模拟温度,通过MD 模拟检测AR-LBD 野生型(AR-LBDWT)和R774C(AR-LBDR774C)的构象变化。结果显示:1)AR-LBDR774C导致局部结构紊乱,从而影响AR-LBD LBP 口袋形状;2)AR-LBDR774C改变AR-LBD 动态性质,进而诱导更多的AR-LBD LBP 构象分布;3)蛋白的构象通过降低温度能够逆转,这与分子生物学实验结果是吻合的。
AR-LBD 二聚体界面间的氨基酸残基突变,是导致AIS 的重要原因之一,而机制尚不清楚。Fu 等[31]构建了2 个代表性的AIS 相关突变AR-LBDW751R和AR-LBDF754V,以及AR-LBDWT的二聚体的MD 模拟,结果显示AR-LBDW751R和AR-LBDF754V二聚体在模拟中非常容易被分离,AR-LBDWT则在模拟过程中始终保持稳定。随后的实验验证支持了模拟结果,即AR-LBDWT能保持二聚体,AR-LBDW751R和ARLBDF754V则以单体形式存在。即AR-LBD 二聚体界面间的突变导致AR 无法二聚化,因此雄激素无法发挥正常功能。
2.2 市售小分子药物耐药机制的研究
市售的AR 拮抗剂,在用药一段时间后,都会发生不同程度的耐药,甚至部分药物会从拮抗剂转变为激动剂。其中,AR 的点突变是最具代表性的耐药机制[3,8]。然而,点突变如何影响受体结构,进而导致药物耐药,这一现象难以从单一的实验现象解释,计算机模拟则给出了重要的补充解释。
R-比卡鲁胺(3)和化合物7 结构高度相似(见图1 和图4)。有趣的是,R-比卡鲁胺在ARLBDWT情况下显示具有拮抗活性,在AR-LBD 点突变Trp741 突变为Leu741(AR-LBDW741L)情况下,则表现为激动活性,类似物7 则无论是否发生ARLBDW741L突变,均表现为激动活性。Bisson 等[32]对化合物3 和7 分别在AR-LBDWT和AR-LBDW741L突变体中进行MD 模拟以探讨机制。结果显示,ARLBDW741L中H3,H5,H11 和H12 的氨基酸残基表现更为稳定,而在AR-LBDWT中,R-比卡鲁胺通过降低氨基酸残基Met895 和Trp741 的稳定性而达到拮抗效果。即Trp741 通过稳定AR 构象在激活状态导致耐药。Osguthorpe 等和Liu 等随后在对R-比卡鲁胺和类似物7 的模拟研究中也得到了类似的结论,即AR-LBDW741L影响H12 的构象是拮抗剂和激动剂变化的关键点[33-34]。除了AR-LBDW741L,Liu等[35]对多种耐药突变W741C,W741L,W741C_T877A,T877A,F876L,F876L_T877A 和L701H的模拟研究显示,这些耐药突变的共同点是会诱导Met895 对R-比卡鲁胺结合的能量贡献增加,进而稳定H12 的构象以形成有利于共激活剂结合的AF2 位点。
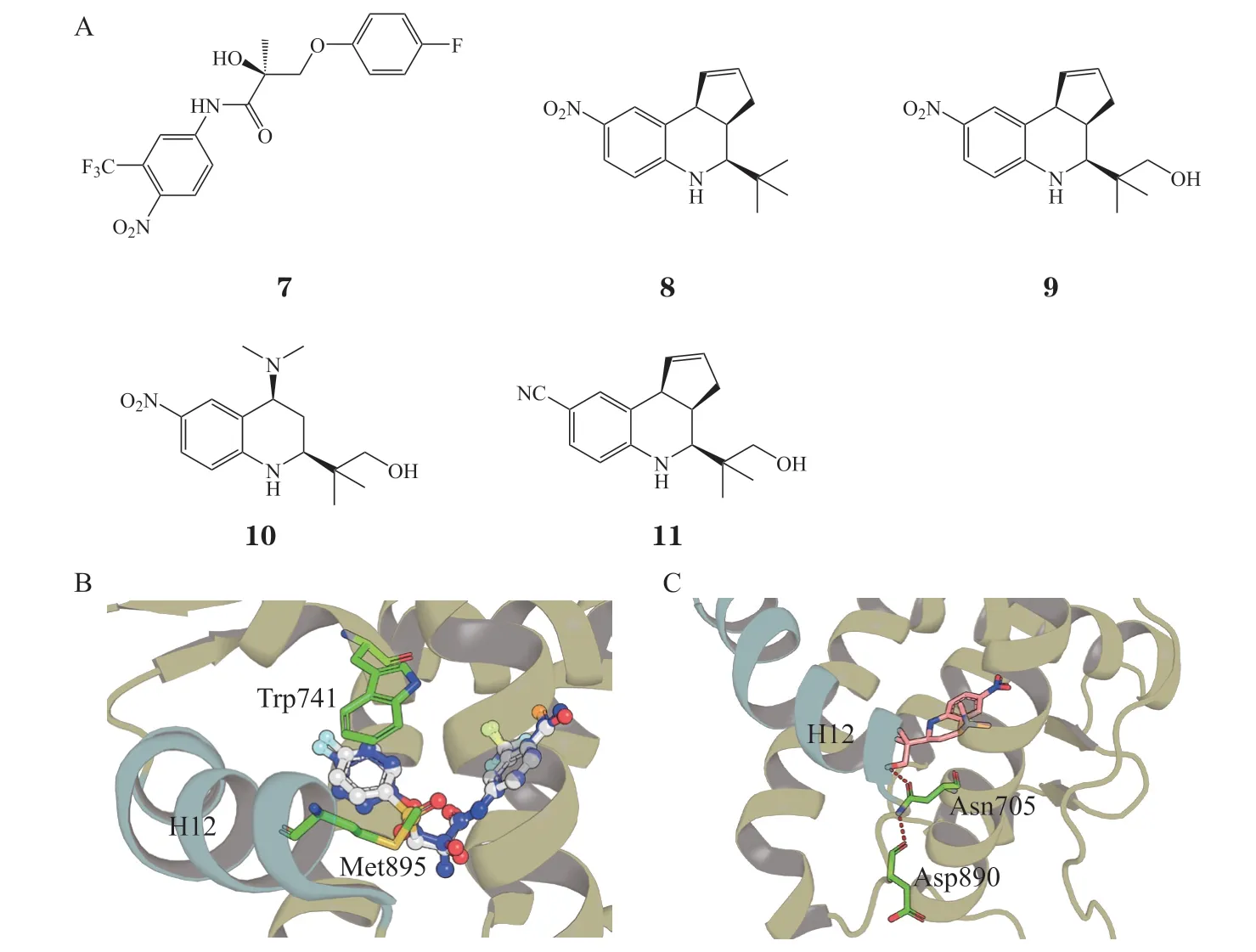
图4 化合物7~11 的化学结构式以及化合物3,7,10 与AR-LBD 的结合模式Figure 4 Structures of compounds 7-11 and the binding modes of compounds 3,7 and 10 with AR-LBD
突变影响AR-LBD H12 构象作为拮抗剂和激动剂转变的关键点,除了在化合物3 和7 中被观察到,在其他拮抗剂中同样有类似的现象。如羟基氟他胺(1,见图1)在AR-LBD 的Thr877 突变为Ala877(AR-LBDT877A)时,会由拮抗剂转变为激动剂。Bisson 等和Liu 等的常规MD 模拟、Zhou 等的副本交换MD 模拟同样显示,AR-LBDT877A突变导致AR-LBD H12 的构象外移,进而影响AF2 位点的构象,是诱导化合物1 转变为激动剂的关键[32,36-37]。
第2 代拮抗剂面临类似问题,如AR-LBD 的Phe876 突变为Leu876(AR-LBDF876L)。由于第2 代拮抗剂与AR-LBD的结合模式类似于第1代拮抗剂,两者的药物转变机制也类似。如Balbas 等和Liu 等的MD 模拟显示,AR-LBDF876L导致恩杂鲁胺(4)的耐药与AR-LBD H12 构象的变化密切相关[38-39]。Gim 等[40]对双氢睾酮(dihydrotestosterone,DHT,天然激动剂)、RU59063(AR 激动剂)以及AR 拮抗剂3~ 5 的加速MD 模拟,也进一步验证了ARLBD H12 的动态波动是拮抗剂和激动剂互相转变的关键。
前述研究在对AR-LBD单体和拮抗剂的模拟中,共同得到了AR-LBD H12 是拮抗态和激动态转变的核心结构这一启示。而随着AR-LBD 的二聚体晶体结构被解析,基于AR-LBD 二聚体的模拟成为可能,Cavaliere 等[41]提出了不一样的耐药机制。他们构建了AR-LBD 二聚体与化合物3 的构象,即每个单体都结合化合物3,2 个单体形成二聚体。MD 模拟研究结果显示,化合物3 结合到AR-LBD 的2 个单体中会导致同源二聚体解离,抑制AR 转录因子活性而发挥拮抗活性;但当AR-LBDW741L突变发生,同源二聚体则具有类似激动剂的行为,即2 个单体紧密结合,这一现象为AR-LBDW741L的耐药机制提供了可能的新的解释。
2.3 微小的结构差异导致截然不同的拮抗/激动活性的机制研究
在AR 的药物设计,特别是AR-LBD LBP 的药物设计中,微小的化学结构差异便会导致截然不同的拮抗和激动活性。为了阐明这一现象的机制,Nagata 等[42]对DHT 和4 个结构相近(四氢喹啉,8~ 11,见图4A),但性质极为不同的ARLBD LBP 配体进行MD 模拟。其中,化合物8 为弱AR 结合,化合物9 和10 对AR 具有高活性激动效应,化合物11 则是高效拮抗剂。特别是相较于化合物8,化合物9 增加了羟基从而极大增强了激动活性。硝基转变为氰基时(化合物9 转变到化合物11),结构相近的分子从激动剂转变为拮抗剂。模拟结果显示,化合物10 的羟基和AR-LBD H3 的Asn705 形成氢键相互作用,而形成的氢键进一步稳定Asp890,进而稳定H12 的构象(见图4C)。拮抗剂11 则是影响H12 的稳定性,达到拮抗效应。
2.4 计算机辅助药物设计技术在环境污染物机制预测中的应用
工业污染、燃烧产物或农药释放所产生的持久性有机污染物(persistent organic pollutants,POPs)在环境中持久存在。POPs 的持续暴露与各种生殖障碍密切相关,如精液质量下降、睾丸癌和性别比例失衡等[43-44]。其中,二氯二苯基二氯乙烯(dichlorodiphenyldichloroethylene,p,p'-DDE,12)和多氯联苯(polychlorinated biphenyls,PCBs)是被最广泛研究的POPs。化合物12 和13(CB-153)是AR拮抗剂(见图5),然而,其拮抗机制尚未得到合理解释。Xu 等[45]采用分子对接、MD 模拟以及基于MM/GBSA 的结合自由能预测研究了化合物12和13 与AR-LBD 的结合模式和拮抗机制。作者对几个潜在结合位点进行了分析,总结出3 个潜在位点,包括AR-LBD 的LBP,AF2 以及N 端的裂口。

图5 化合物12~ 17 的化学结构式Figure 5 Structures of compounds 12 -17
溴系阻燃剂如多溴联苯醚(polybrominated diphenyl ethers,PBDEs)是家用和工业产品中常用的添加剂,由于这些添加剂长期暴露问题,其代谢产物的毒性问题备受关注[46]。PBDEs 最常见的代谢产物14 [(羟基-PBDEs(HO-PBDEs)和甲氧基-PBDEs(MeO-PBDEs),见图5],可在人类血液、海洋生物和野生动物中等被检测到[46-48]。研究显示HO-PBDEs和MeO-PBDEs 能够抑制AR 活性,然而其抑制机制尚不清楚[49-50]。Wang 等[50]通过细胞水平实验检测、分子对接和MD 模拟相结合研究了多种HO-PBDEs和MeO-PBDEs 抑制AR 活性的机制。结果显示,ARLBD H12 构象的变化是PBDEs 代谢产物具有拮抗活性的重要指标,为评估PBDEs代谢产物提供了预测模型。
化合物15(二苯甲酮-1,benzophenone-1,BP-1,见图5)是常用的紫外线过滤材料之一,广泛应用于化妆品和防晒霜等个人护理品中。BP-1 残留物在多种水体如泳池水中被频繁检出,对内分泌有一定影响但作用机制尚不明确。为此,Zhan 等[51]研究了BP-1 在泳池中的氯化过程,并通过高斯加速MD 模拟评估了BP-1 氯化产物对AR-LBD 的影响。体外实验结果显示BP-1 在泳池中的氯化产物16(一氯代BP-1)和17(二氯代BP-1)(见图5)对AR具有抑制作用;MD 模拟显示两者通过疏水和非极性相互作用与AR 稳定结合,表现出强于化合物15的拮抗活性。研究结果提示个人护理产品会阻断AR 信号通路,进而具有潜在的健康风险。
2.5 计算机辅助药物设计技术在共激活剂机制阐明中的应用
共激活剂与AR-LBD 的结合是AR 调控男性发育过程中必不可少的环节,不同组织中不同的共激活剂与AR-LBD 的结合导致AR 的构象变化进而发挥不同的生理功能[52]。然而,共激活剂和受体两者结合的动态激活机制尚未明确阐明。
为了探讨共激活剂和激动剂协同结合AR-LBD的机制,Xu 等[53]构建了AR-apo(无配体的ARLBD)、AR 结合DHT、AR 结合类固醇受体共激活剂(steroid receptor coactivator,SRC)、AR 结合DHT 和SRC 这4 个不同的结构。通过MD 模拟探讨共激活剂和激动剂在AR 转录中潜在的相互作用模式。模拟结果显示:1)当共激活剂结合AR-LBD时,会增加AR-LBD 结构的动态稳定性,并诱导增大AR-LBD LBP 位点的体积,促进激动剂的结合(见图6A);2)类似地,当激动剂结合到AR-LBD 时,会引起AF2 位点区域的重排,进而诱导出适合共激活剂结合的结合区域(见图6B)。总的来说,激活剂和激动剂两者结合AR-LBD是双向作用,互相促进。
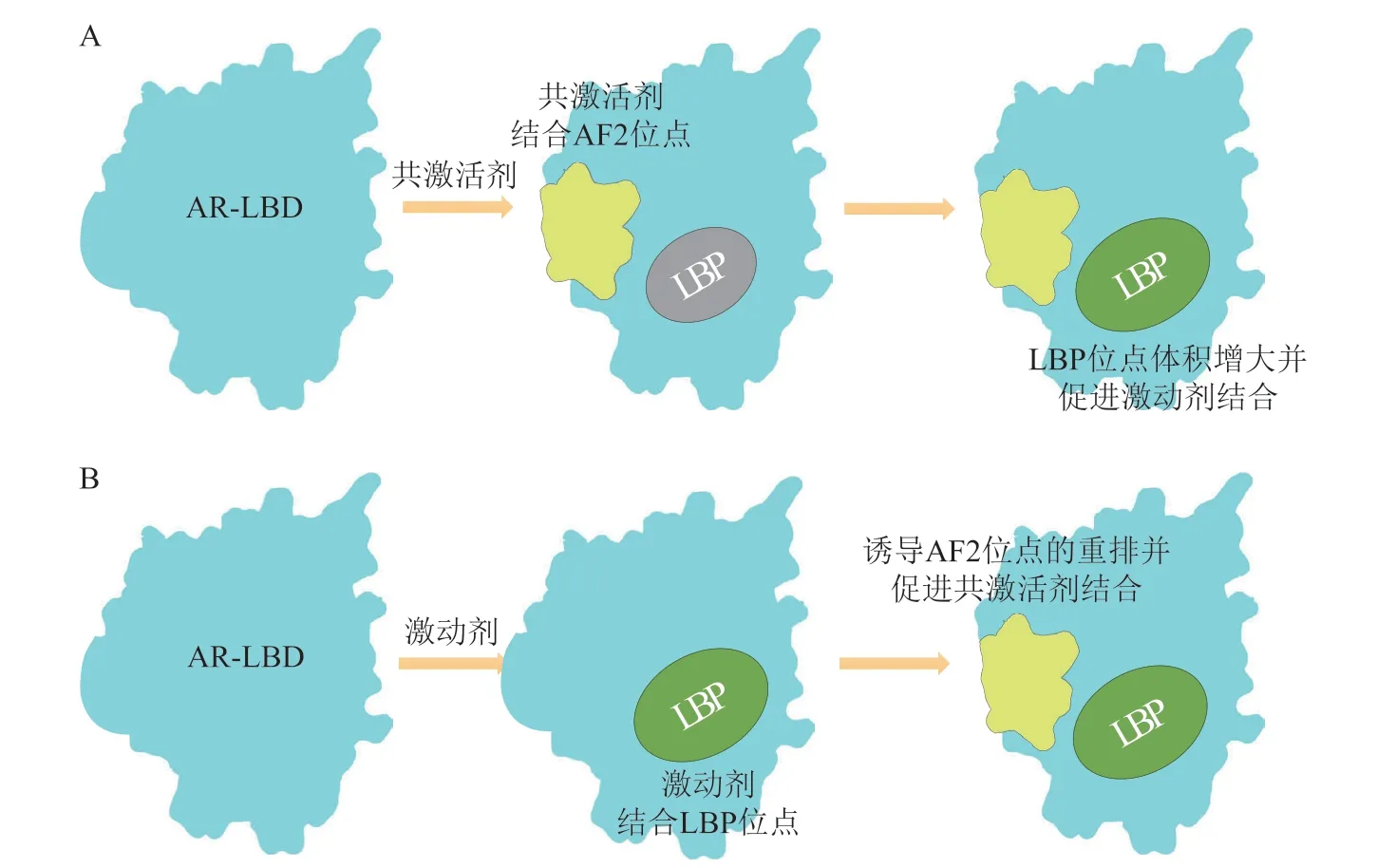
图6 共激活剂和激动剂协同结合AR-LBD 的机制示意图Figure 6 Schematic diagram of the dynamic communication between coactivators and agonists bound to AR-LBD
为了探讨激动剂和拮抗剂与AR-LBD 结合后对AR-LBD 与共激活剂结合的影响,Liu 等[54]对多种共激活剂和多种拮抗剂与AR-LBDWT和ARLBDF876L进行了MD 模拟,结果表明不同激动剂和拮抗剂结合AR-LBD 会调控与AF2 位点相关的配体-共激活剂变构路径。自由能计算表明,共激活剂与AR-apo 之间的结合亲和力大于AR 结合拮抗剂,小于AR 结合激动剂。此外,AR-LBD 的F876L 突变影响配体-共激活剂的变构途径,可能是点突变诱导拮抗剂(如化合物4)耐药的重要原因。Jin 等对AR-LBD 结合激动剂、拮抗剂以及共激活剂的模拟与Xu 等和Liu 等的模拟结果类似[53-55]。激动剂的结合促进了AF2 位点的构象形成,拮抗剂则干扰AF2 位点的形成,进而减弱与共激活剂结合[55]。
研究发现,新型的共激活剂Leupaxin(LPXN)与AR 相互作用,并在前列腺癌的侵袭和进展中发挥重要作用[56]。Khan 等[57]对LPXN 与AR-LBDWT以及临床常见的突变AR-LBDH874Y,AR-LBDT877A和AR-LBDT877S的相互作用进行了MD 模拟研究。结果表明,LPXN 能与AR-LBDWT以及突变体稳定结合,并且结合自由能计算结果显示AR-LBDWT为(-32.95±0.17)kcal·mol-1,而AR-LBDH874Y,ARLBDT877A和AR-LBDT877S分别为(-36.69±0.11),(-38.78±0.17)和(-41.16±0.12)kcal·mol-1,表明这些突变潜在增加了LPXN 的结合,从而促进前列腺癌侵袭和迁移。
3 计算机辅助药物设计技术在发现雄激素受体新位点、新母核方面的应用
CADD 技术除了在机制探索方面被广泛应用,在寻找靶标新位点、新母核方面也具有独特的优势[58-59]。靶标新位点的发现、鉴定以及全新分子的发现,通常需要消耗大量的人力和物力。传统模式一般是通过生物实验对实体化合物库进行高通量筛选,以发现苗头分子。这种模式虽能有效发现苗头分子,但会消耗极大量的资源,并存在高假阳性率问题。随着靶标三维结构被不断解析,基于CADD 技术以及融合AI 技术的结合口袋探测、基于受体(或配体)的高通量虚拟筛选的应用,为制药行业,特别是新分子发现,带来了前所未有的变革[59-61]。
如表1 所示,仅对于AR 这一靶标,就有多个位点和新型母核小分子(18~ 41)的发现与CADD 技术密切相关。以下就CADD 技术在AR多个位点中的“首次应用”加以重点描述,以更好地阐述CADD 技术在AR 新位点、新分子发现中的重要作用。
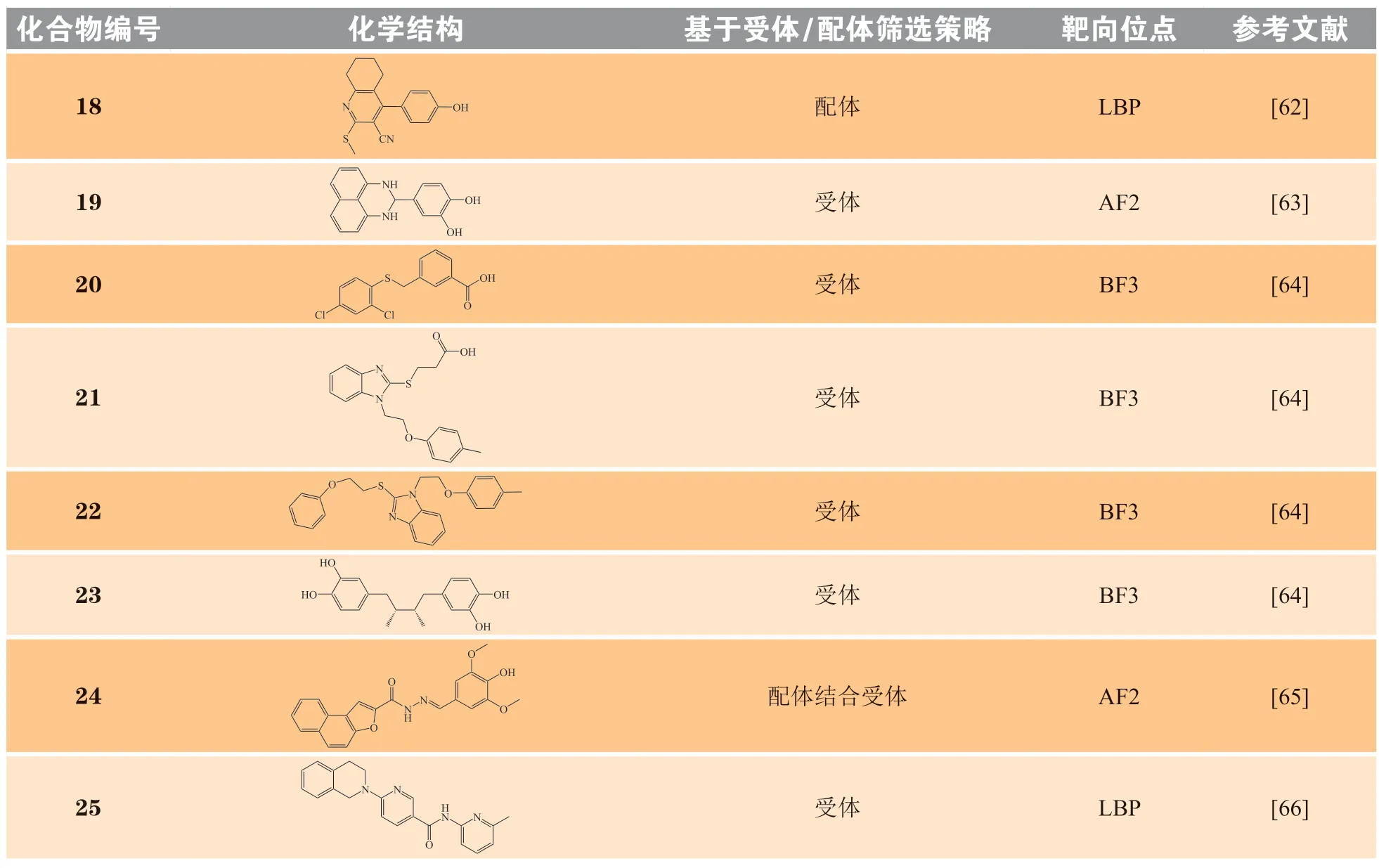
表1 通过计算机辅助药物设计技术发现的靶向雄激素受体的小分子拮抗剂Table 1 The small-molecule antagonists of androgen receptor discovered by computer-aided drug design
Axerio-Cilies 等[63]在2011 年报道了通过高通量虚拟筛选发现AR-LBD AF2 位点拮抗剂的研究结果。通过对约400 万的ZINC 类药库进行层级式虚拟筛选、共识打分结合投票(结合多种打分技术,构象分析等预测技术共同优选候选分子)、人工挑选相结合,不断缩小候选分子的范围,最终选择68个分子进行活性评价。实验验证结果显示,6 个化合物具有抑制AR 转录活性的作用。其中,化合物19 通过与AR-LBD 共晶(PDB 编号:2YHD)验证了筛选的结果。
BF3 位点的发现最早源于高通量筛选发现的苗头分子,然而,靶标位点亲和力不足的问题,促使更多的研究采用不同策略发掘新型母核和具有更高亲和力的分子[64,81]。Lack 等[64]采用与Axerio-Cilies等[63]类似的虚拟筛选策略,最终挑选了213 个候选分子进行活性测试,其中化合物20~ 23 通过与AR-LBD 共晶(PDB 编号:2YLP,2YLQ,2YLO,3ZQT)验证了筛选的结果。
除了在已经发现靶标位点的实验基础上通过高通量虚拟筛选发现新型母核,在实验没有发现的新位点时,通过CADD 技术,发现潜在位点,并对发现的新位点进行高通量虚拟筛选发现新型候选分子是CADD 技术高效优势的重要体现。Li 等[70]通过Molecular Operating Environment 软件的Site Finder模块对AR-DBD 区域进行了深入探索,发现ARDBD 的P-box 区域与DNA 结合区域间存在1 个结合口袋(见图2A)。接着,通过高通量虚拟筛选以及对苗头分子的结构优化,最终得到化合物29。由于与上市药物恩杂鲁胺不在同一结合区域,因此化合物29 能够有效抑制恩杂鲁胺耐药细胞株的增殖。除了在P-box 区域发现候选分子,Dalal 等[72]在AR-DBD 的D-box 发现存在小分子结合位点(见图2A),并在该区域通过高通量虚拟筛选发现候选分子31(结构未公布),该化合物能够阻断AR-DBD的二聚化,并抑制AR 转录活性。
除了在AR-DBD 区域利用CADD 技术发现新位点、新分子,Fu 等[31]通过CADD 技术发现了AR-LBD 区域的第4 个新位点(DIP 位点,见图2),并在此基础上,发现了全新的高活性候选分子。他们通过MD 模拟和实验验证确定了AR-LBD 二聚体易被破坏,而如果有小分子阻断AR-LBD 二聚化,则可能是靶向治疗前列腺癌的新策略。幸运的是,他们发现了二聚体界面间存在1 个较大的空腔,能够容纳小分子。随后,他们对商业化合物库进行了高通量虚拟筛选,并在发现苗头分子的基础上进行结构修饰,最终得到具有进一步开发潜力的候选分子41。
4 结语与展望
CADD 技术在现代新药研发中,特别是在前期发现阶段,在机制研究、靶标新位点、新母核发现方面,相比经典实验方式具有高效、经济的优势。然而,CADD 技术在不同研发阶段,需要使用不同的方法和技术。例如,在靶标新位点发现方面,MD 模拟结合新口袋检测技术;在动态检测新位点方面,相比经典实验更易找到一些变构位点以及隐藏的位点;在位点新母核发现方面,基于受体/配体的发现方式,结合多种评分机制能够有效筛选大量的化合物数据库,提高苗头分子发现的效率;在经典位点发现新母核分子方面,最新的基于大数据的分子生成技术,能够快速生成性质相似,但母核结构不一样的分子,为快速获得新母核提供了技术支持;在已有苗头分子的基础上,CADD 技术也提供了相关的技术方案,包括高精度自由能计算方法(热力学积分和自由能微扰等),以及基于AI 的ADMET 预测工具,为成药性开发提供了技术支持。总之,CADD 技术贯穿新药研发的早期研发阶段,是现代新药开发不可或缺的辅助技术。随着CADD技术的不断发展和实践应用,该技术对包括AR 在内的多个传统靶标,以及一些新兴潜力靶标,在未来新药开发中必将发挥举足轻重的作用。
