鞘胺醇-1-磷酸受体1 选择性调节剂的研究进展
2023-10-20王茜茜江程
王茜茜,江程
(中国药科大学药学院,江苏 南京 211198)

近年来,鞘氨醇-1-磷酸(sphingosine-1-phosphate,S1P,1)受体调节剂作为一类新型免疫抑制剂受到广泛关注,它通过影响机体免疫应答和免疫病理反应而抑制机体异常免疫反应,在临床上主要用于自身免疫性疾病以及器官移植排斥反应的治疗。
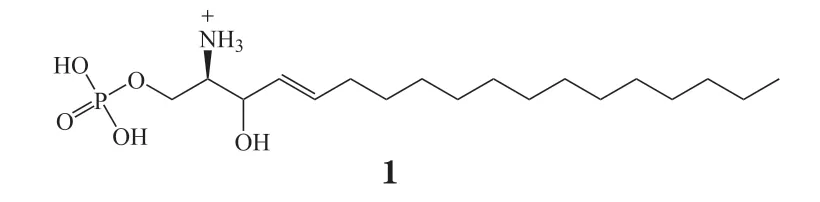
S1P 为简单的膜衍生溶血磷脂[1],来源于鞘氨醇(sphingosine,Sph,大多数鞘脂的主干)[2]。鞘氨醇由软脂酰辅酶A 及丝氨酸在磷酸吡哆醛、还原型辅酶Ⅱ及黄素腺嘌呤二核苷酸等辅酶参与下合成,也可由神经酰胺在神经酰胺酶作用下脱酰胺键生成。鞘氨醇合成的关键酶是鞘氨醇激酶(sphingosine kinase,SphK),Sph 在SphK 催化下与磷酸通过磷酸酯键相连生成S1P[3]。SphK 也是限速酶,从而调节S1P 的体内平衡。在哺乳动物中,SphK 存在SphK1 和SphK2 亚型,这2 种亚型由不同基因转录和翻译生成,其基因具有高度同源性[4]。
1 鞘氨醇-1-磷酸受体的分类及生物学功能
S1P 通过与5 种已知的G 蛋白偶联鞘氨醇-1-磷酸受体(sphingosine-1-phosphate receptor,S1PR)中的任何1 种结合来调节一系列细胞过程,这5 种受体亚型通过激动各自的细胞内信号途径而发挥作用[5]。S1PR1主要与Gi 蛋白偶联以抑制环磷酸腺苷(cyclic adenosine monophosphate,cAMP)的产生;S1PR2和S1PR3与Gi,Gq 和G12/13 偶 联;S1PR4和S1PR5则通过Gi 和G12/13 发出信号[6]。尽管S1PR 中各亚型的蛋白序列高度保守,但其结构仍有不同,如S1PR 中被称为ICL2 和ICL3 的2 个区域显示出相当大的结构和序列多样性,这些区域在单个受体中的动态构象对G 蛋白偶联特异性有贡献[7];S1PR1和S1PR3的结构显示有1 个通向外部的空腔,然而,类似该空腔的位置在S1PR2中是闭合的[8];与S1PR1-Gi 和S1PR5-Gi 复合物结构的比较表明,S1PR3中的ICL2 区域折叠成明显的延伸构象[9]。各亚型结构和序列的不同是造成其功能不同的主要原因,同时也为研究具有选择性的S1PR 调节剂提供了重要依据。
1.1 S1PR1
S1PR1可以调控胸腺、次级淋巴器官和骨髓中的淋巴细胞排出,起到调节免疫的作用。这种调节作用依赖于S1P 浓度变化,在组织中S1P 裂解酶降解S1P 使S1P 浓度降低,而外周血中不存在这种作用,从而产生了从组织到血液/淋巴的S1P浓度梯度,淋巴细胞依靠在细胞表面表达的S1PR1,通过感受S1P 浓度的变化,从淋巴结和次级淋巴器官中移出,使血液中的淋巴细胞浓度增加,增强了免疫作用。然而,当淋巴细胞表面的S1PR1受到调节剂的刺激时会被细胞内吞作用迅速内化,淋巴细胞就会失去感受S1P 浓度的能力,而被淋巴器官“扣留”,不能够进入淋巴循环,从而降低了血液中淋巴细胞的浓度,起到了免疫抑制作用[10-11]。S1PR1选择性调节剂就是通过这种机制来达到调控淋巴细胞的作用,对多发性硬化症(multiple sclerosis,MS)[12]、炎性肠病(inflammatory bowel disease,IBD)[13]及移植物抗宿主病(graft-versus-host disease,GVHD)[12]等免疫相关疾病具有潜在治疗作用。
除调节免疫的作用外,S1PR1也具有参与树突状细胞募集、调节血管通透性[14-15]、参与星形胶质细胞增殖、保护神经、调节心率、保护内皮完整性[16-17]等作用。研究表明,在人体内S1PR1选择性调节剂具有通过激活G 蛋白门控内向整流钾离子通道(G protein gated inwardly rectifying K+channels,GIRKs)而导致心动过缓的副作用[18-19]。由此可见,这些不同的生物学效应是各种疾病的潜在治疗靶点,也可能产生脱靶的不良反应。
1.2 S1PR2 和S1PR3
S1PR2可能具有与S1PR1相反的功能[20],具有促炎作用。S1PR2和S1PR3介导了血管、肠、支气管和膀胱平滑肌等收缩反应[21]。对啮齿类动物的研究表明,S1PR 调节剂引起心动过缓是由于对S1PR3的激动作用[22]。且有研究表明,S1PR3与高血压有关[23],S1PR2和S1PR3与促纤维化反应相关[24]。此外,S1PR2和S1PR3还参与了一些病理生理过程,包括炎症、腹胀、癌细胞生长和血管生成[21]。
1.3 S1PR4 和S1PR5
目前对于S1PR4的了解最少,但已知S1PR4在免疫细胞迁移和某些免疫细胞的分化中发挥作用[25-26]。S1PR5可使自然杀伤细胞从骨髓和淋巴结转移到组织中[27]。此外,它还能调节脑内皮屏障的功能、紧密连接和通透性[28],并通过降低黏附分子、炎症性趋化因子和细胞因子[26]的表达来降低脑内皮细胞上核因子-κB(nuclear factor-κB,NF-κB)的激活。然而,目前尚不确定S1PR1调节剂与S1PR5的相互作用是否有助于S1PR1调节剂发挥对MS 的临床疗效。
2 鞘氨醇-1-磷酸受体1 调节剂
作为新兴的口服免疫抑制剂,目前已有4 个S1PR1调节剂上市,多个S1PR1调节剂处于临床研究阶段(见表1)。以下按化学结构分类介绍各S1PR1调节剂的研究进展。
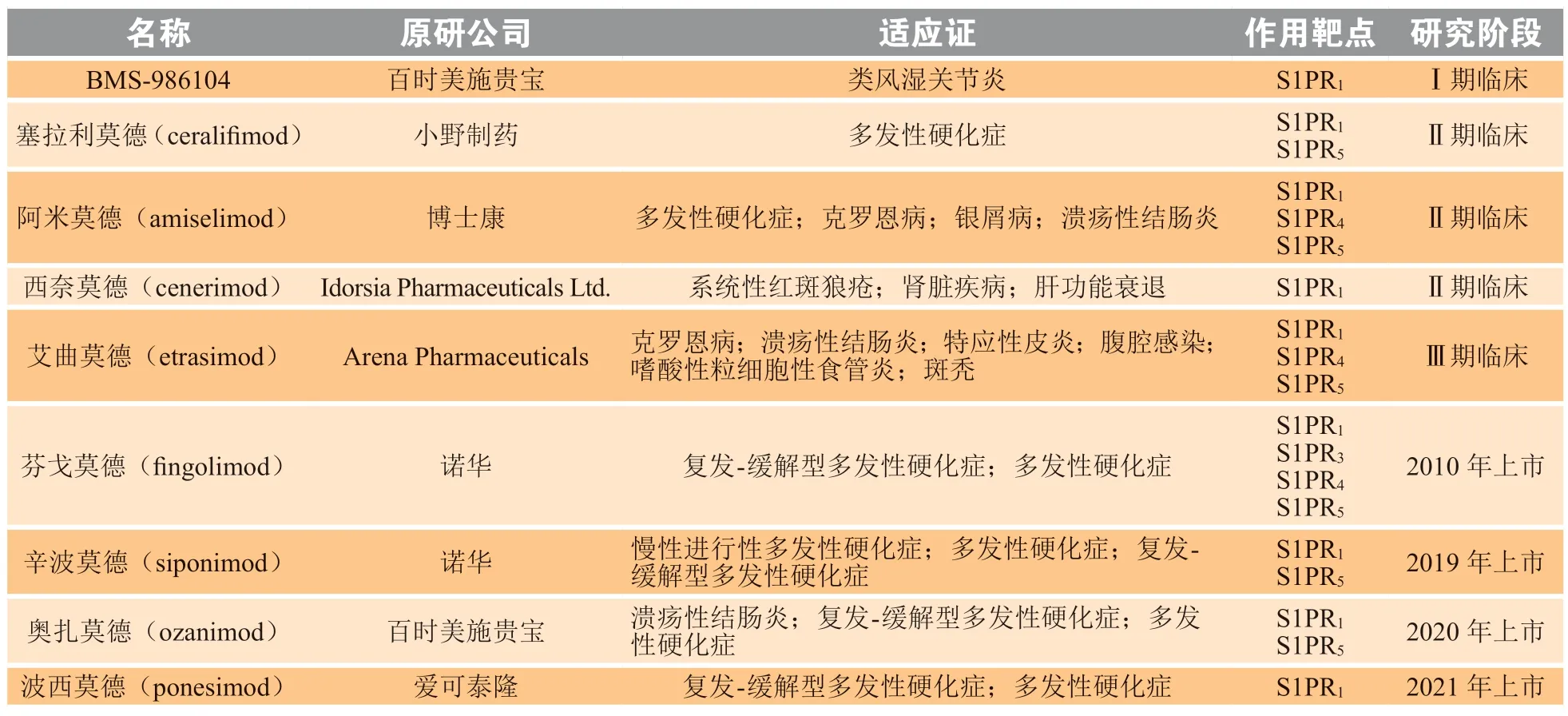
表1 鞘氨醇-1-磷酸受体1 选择性调节剂临床研究进展与上市情况Table 1 Progress of clinical research on and marketing of sphingosine-1-phosphate receptor 1 selective modulators
2.1 氨基醇类化合物
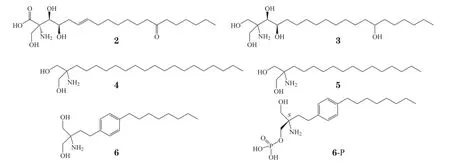
2.1.1 芬戈莫德在S1PR1激动剂出现之前,MS 患者的药物治疗选择包括免疫调节剂、免疫抑制剂和抗炎药,如醋酸格拉替拉末、环孢素A、他克莫司(FK506)、米托蒽醌等,这些药物虽有一定的疗效,但具有安全性差、不良反应多的缺陷。20 世纪80 年代末至90 年代初,Kiuchi 等[29]在真菌Isaria sinclaiii的提取物中分离出具有免疫抑制活性的化合物ISP-I(2)。小鼠同种异体混合淋巴细胞反应(mixed lymphocyte reaction,MLR)试验结果显示,ISP-I 的抑制活性是环孢素A 的5~10 倍(IC50分别为3~ 8 和14 nmol·L-1),但ISP-I 在1.0 mg·kg-1的剂量下对大鼠有毒性作用,而环孢素A 在100 mg·kg-1时才产生毒性,且ISP-I 溶解性差。为了改善ISP-I 的安全性和溶解性,他们对ISP-I 进行了一系列结构修饰:首先将ISP-I 的双键还原成单键,并将羧酸和酮羰基还原成醇,得到毒性降低且可溶性提高的ISP-I-28(3),但ISP-I-28 的抑制活性相对较弱(IC50=1 630 nmol·L-1);进一步将ISP-I-28结构中3 个羟基去除,留下1 个18 碳烷基链,得到化合物ISP-I-36(4),其抑制活性提高(IC50=12 nmol·L-1);再将ISP-I-36 的烷基链从18 个碳缩短到14 个碳,得到ISP-I-55(5),抑制作用进一步加强(IC50=5.9 nmol·L-1);最后通过限制构象来降低毒性并提高活性,引入1 个芳香族部分,得到理化性质更理想、毒性更小的芬戈莫德(fingolimod,6,IC50=6.1 nmol·L-1)。
芬戈莫德由诺华制药公司研制而成,2010年美国食品药品监督管理局(Food and Drug Administration,FDA)批准该药以商品名Gilenya上市,作为一线药物治疗复发型MS。Gilenya 是首个适用于复发型MS 的口服治疗药物。一旦摄入,即被SphK2 迅速磷酸化,形成芬戈莫德-P(6-P)。芬戈莫德-P 作用机制的新颖之处在于该化合物能够重新分配血液中循环的淋巴细胞,而不会减少淋巴细胞总数。芬戈莫德-P 对S1PR1的结合亲和力最高(EC50=0.087 nmol·L-1),对S1PR3,S1PR4和S1PR5的亲和力稍低(EC50分别为5.1,66.3 和0.24 nmol·L-1),对S1PR2则没有亲和力[30]。
芬戈莫德-P 与S1PR1结合后会导致S1PR1内化和降解,导致细胞表面的S1PR1下降,细胞表面S1PR1恢复需要较长的时间,这使得芬戈莫德具有长时间的免疫抑制作用[31]。在体内活化后的芬戈莫德-P 含有2 个负电荷,容易与体内蛋白发生非特异性结合,导致半衰期过长、分布容积过大。而且芬戈莫德对S1PR1,S1PR3,S1PR4和S1PR5均具有激动作用,选择性较低,从而导致感染、神经系统紊乱、胃肠道疾病、血管收缩、支气管收缩等不良反应[32]。目前,正在进行芬戈莫德用于消除肾移植后间质纤维化和肾小管萎缩的Ⅱ期临床试验(临床试验编号:NCT05285878)和考察芬戈莫德(Gilenya,0.5 mg)在中国复发型MS 患者中的疗效和安全性 的Ⅳ期临床试验(临床试验编号:NCT04667949)等。
2.1.2 阿米莫德传统的2-氨基丙烷-l,3-二醇化合物会造成短暂的心动过缓,为了解决这个问题,Kiuchi 等[33]决定对氨基丙烷头部和长烷基链尾部进行改造,并在苯环上引入其他取代基以提高选择性,最终得到阿米莫德(7)。
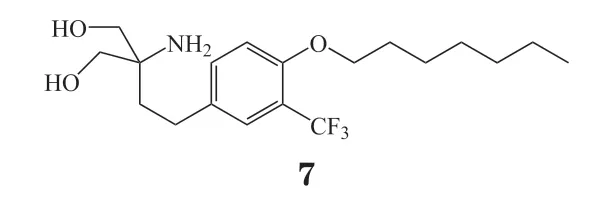
与芬戈莫德一样,阿米莫德在体内磷酸化为阿米莫德-P 后发挥作用。阿米莫德是第2 代S1PR1调节剂,具有优异的免疫抑制和排斥反应抑制效果等。在GIRK 活性测试中,阿米莫德-P 的活性仅为芬戈莫德-P 活性的1/5,表明阿米莫德-P 减少了心动过缓的不良反应;在对人S1PR 激动活性的测试中,阿米莫德-P 对S1PR1和S1PR5都有较好的选择性(EC50分别为0.075 和0.47 nmol·L-1),对S1PR4的激动作用较弱(EC50=122.3 nmol·L-1),对S1PR2或S1PR3则无明显的激动作用(EC50>10 000 nmol·L-1)[30]。目前,正在进行一项考察阿米莫德治疗轻中度溃疡性结肠炎的疗效和安全性的Ⅱ期临床试验(临床试验编号:NCT04857112)。
2.1.3 VPC 01091 和BMS-986104仅消除对S1PR3的激动作用不足以消除芬戈莫德或选择性S1PR1全激动剂在啮齿类动物中所引起的急性和慢性肺毒性。Zhu 等[34]以芬戈莫德为模板,通过限制氨基取代碳“头基”和辛基苯基“尾基”的构象,得到化合物VPC 01091(8)。该化合物在啮齿类动物中引起了明显的、持久的淋巴细胞减少,而未导致心率降低。VPC 01091 有4 种光学异构体,其中8a 和8b 被磷酸化后是S1PR1受体的有效部分激动剂(EC50分别为6.56 和5.17 nmol·L-1)。VPC 01091 尚未进入临床研究。
基于对VPC 01091 的研究,Dhar 等[35]决定探索功能性构象上以四氢萘环系统的形式限制VPC 01091的侧链的效果,得到BMS-986104(9)。在鸟苷5'-O-(3-硫代三磷酸)[guanosine 5'-[γ-thio]triphosphate,GTPγS]结合试验中,BMS-986104-P 对S1PR1显示出较强活性(EC50=0.901 nmol·L-1),而对S1PR3未显示出活性。此外,BMS-986104 对心血管和肺有较好的安全性,且在T 细胞转移性结肠炎模型中与化合物3 显示出同等的疗效。目前已完成一项评估BMS-986104 在健康男性受试者中的安全性、耐受性、药代动力学和药效学的随机、安慰剂对照、双盲、单次给药剂量递增Ⅰ期临床试验(临床试验编号:NCT02211469)。
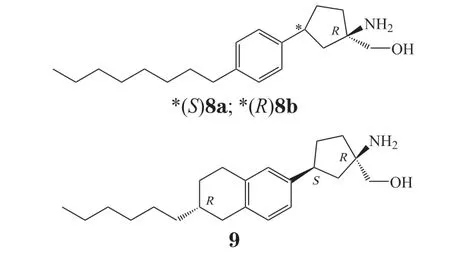
2.1.4 CS-0777已有研究表明,以五元杂环为连接臂的化合物可能比以苯环为连接臂的化合物对S1PR1有更高的选择性,日本Daiichi Sankyo 公司设计出结构新颖的S1PR1选择性调节剂CS-0777(10)[36]。在GTPγS 结合试验中,观察到CS-0777-P(体内磷酸化产物)对人S1PR1具有较强的激动作用(EC50=1.1 nmol·L-1),对S1PR3的 激动作用则较弱(EC50=350 nmol·L-1)。大鼠模型试验表明,CS-0777 可降低外周血淋巴细胞,并能抑制试验性过敏性脑脊髓炎(experimental allergic encephalomyelitis,EAE)的发展,且有良好的药代动力学性质[37]。目前已完成MS 患者口服CS-0777的12 周安全性评估的Ⅰ期临床试验(临床试验编号:NCT00616733)。
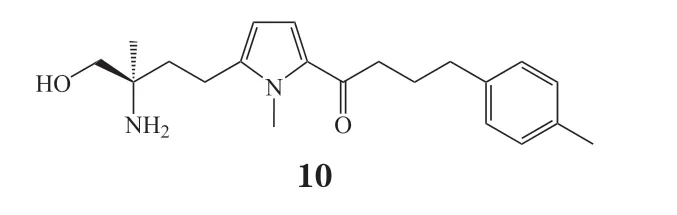
2.1.5 奥扎莫德由于使用芬戈莫德治疗后需要超过5 周淋巴细胞才能完全恢复,可能增加感染的风险,因此需要寻找消除半衰期短的新药物[38]。研究表明3,5-二芳基-1,2,4-二唑类化合物对S1PR1表现出良好的激动作用,对S1PR3的选择性也进一步提升[39]。奥扎莫德(11)是以3,5-二芳基-1,2,4-二唑为骨架设计而成的小分子S1PR1和S1PR5选择性激动剂(EC50分别为0.41 和11 nmol·L-1)。该药口服有效,具有较好的受体选择性和良好的药代动力学性质,包括循环半衰期短、淋巴细胞可以快速再增殖[40],以及避免因长时间的淋巴细胞减少而造成的免疫功能下降。与芬戈莫德相比,奥扎莫德的安全性有所提高,然而,由于S1PR1存在于心房肌细胞上,首次给药后仍可能出现心脏副作用,但仅限于治疗开始时心率的短暂降低,可通过滴定疗法得到缓解[41]。目前,正在进行的临床试验包括奥扎莫德对新型冠状病毒感染的干预研究(Ⅱ期,临床试验编号:NCT04405102)和口服奥扎莫德治疗中重度活动性克罗恩病的扩展研究(Ⅲ期,临床试验编号:NCT03467958)等。
2.2 氨基羧酸类化合物
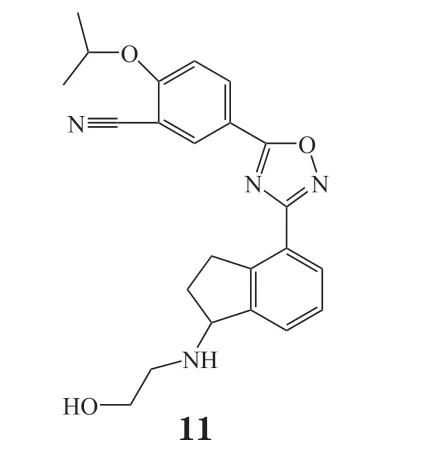
2.2.1 辛波莫德Pan 等[42]设想在芬戈莫德的亲脂性烷基链中增加刚性的修饰可能会提高S1PR 亚型选择性,发现了含有取代烯丙基部分的取代苄氧基肟的类似物12。小鼠模型试验结果显示,经口给药时,化合物12 在诱导淋巴细胞再分配方面与芬戈莫德等效。然而,其相应磷酸盐的体内消除半衰期仍然很长(t1/2>30 h),且分布体积很大(Vss=36.9 L·kg-1),造成这种药代动力学行为的原因可能是体内产生的带电荷的磷酸部分与组织中脂蛋白高度非特异性结合。为此,Pan 等[42]决定用羧酸取代磷酸部分以减少非特异性结合,从而减少分布体积并缩短消除半衰期。为了有效地确定最佳羧酸基团,他们采用还原胺化策略,将不同的氨基羧酸与化合物12 的“疏水尾”对应的醛片段进行结合,得到具有不同羧酸头部的化合物,其中活性最好的为化合物13,且在GTPγS 结合试验中,化合物13对S1PR1的EC50为300 nmol·L-1。将该化合物作为新先导化合物,进一步修饰羧酸头部,最终得到辛波莫德(14)。
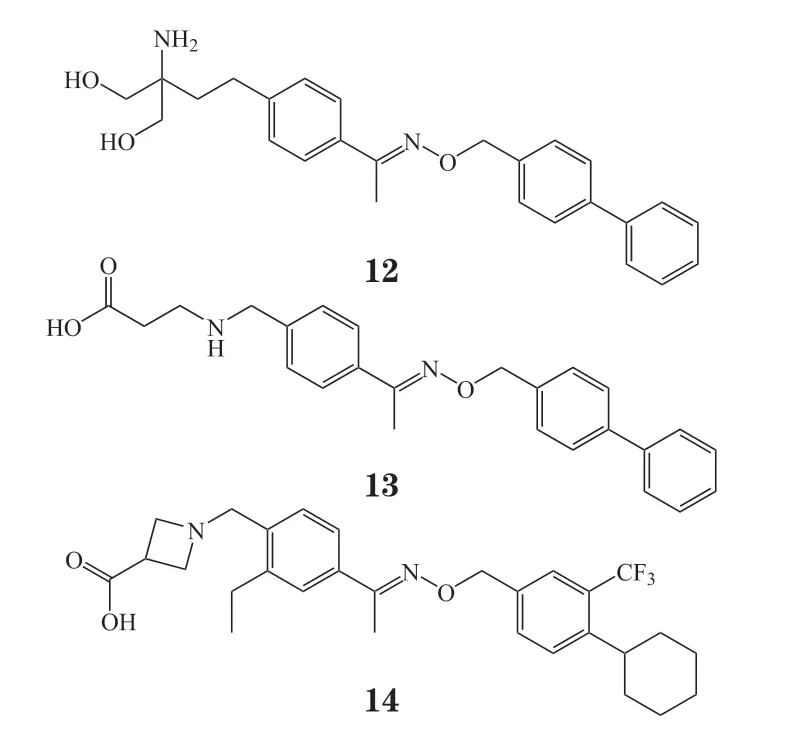
2019 年3 月26 日,美国FDA 批准辛波莫德以Mayzent 为商品名上市,用于治疗成人复发型MS,包括临床孤立综合征(clinical isolated syndrome,CIS)、复发缓解型MS(relapsing-remitting multiple sclerosis,RRMS)和继发进展型MS(secondary progressive multiple sclerosis,SPMS)。Mayzent 是首款获FDA 批准的能有效缓解SPMS 的药物。辛波莫德是新型烷氧基亚氨基衍生物,是S1PR1(EC50=0.39 nmol·L-1)和S1PR5(EC50=0.98 nmol·L-1)的选择性激动剂,在选择性、安全性和药代动力学性质等方面均有明显的优势。与需要在体内转化为磷酸化活性代谢物的前药相比,直接作用的S1PR1激动剂可更准确地控制其体内药理活性[42]。目前,正在进行的临床试验包括评估Mayzent 对SPMS 患者小胶质细胞疗效的研究(Ⅳ期,临床试验编号:NCT04925557),以及考察奥法木单抗和辛波莫德(阳性对照药:芬戈莫德)分别在儿童MS 患者中的疗效和安全性的研究(Ⅲ期,临床试验编号:NCT04926818)等。
为了探究激动剂配体激活受体的机制以及内源性配体和治疗性配体之间的构象差异,Xu 等[43]研究了S1PR1与平衡配体和偏爱性配体的复合物的结构。一般来说,G 蛋白偶联受体的配体可以结合并激活G 蛋白和β-arrestin(β-arrestin 是一类介导受体脱敏的重要可溶性蛋白质,对绝大部分与受体偶联G 蛋白介导的信号转导具有重要调节作用[44])两条途径,优先结合其中之一而不是都结合的药物可以在保留一条途径的有益作用的同时,选择性地避免另一条途径所引起的不必要的副作用。S1P、芬戈莫德-P 和辛波莫德3 个化合物与S1PR 结合的晶体结构如图1 所示(黄色虚线为氢键)。与S1P 和芬戈莫德-P相比,辛波莫德的柔性烷基链被刚性苄氧基肟部分取代,磷酸头被氨基羧酸取代,其结合结构的配体密度(ligand density)最好。在配体结合口袋中,最显著的差异是关键氨基酸W269 的位置和取向,其翻转会激活S1PR1并触发β-arrestin 途径。从配体结构的角度来看,β-arrestin 偏爱性配体芬戈莫德-P 和辛波莫德都有较短的尾部和较大的中间部分,二者距W269较S1P 更近,优先与W269 互动;同时,它们较大的中间部分可能有助于增加配体口袋的大小,以促进W269 与关键残基的相互作用并最终激活β-arrestin 途径,是有利于β-arrestin 相互作用的配体构象。
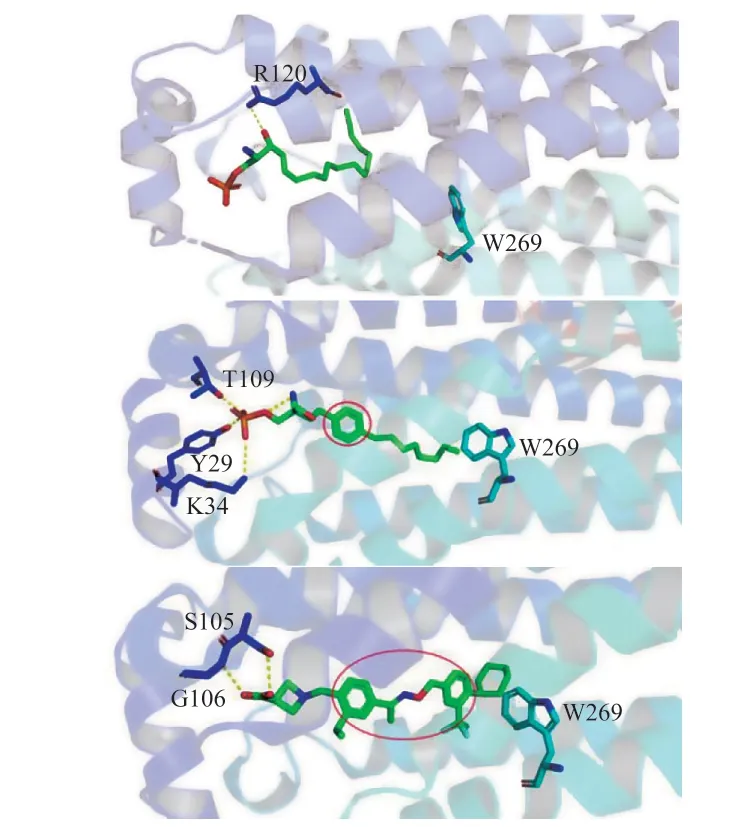
图1 鞘氨醇-1-磷酸、芬戈莫德-P 和辛波莫德分别与鞘氨醇-1-磷酸受体1 结合的蛋白晶体结构Figure 1 Protein crystal structures of sphingosine-1-phosphate,fingolimod-P and siponimod binding,respectively,to sphingosine-1-phosphate receptor 1
2.2.2 AMG 369 及类似苯并噻唑类化合物自苯丙酸类S1PR1选择性激动剂问世后,科学家又先后开发出了氮(杂)环丁烷-3-羧酸衍生物,大多数氮(杂)环丁烷-3-羧酸衍生物对S1PR1/S1PR5基本上没有相对选择性。Frohn 等[45]在对苯并环化化合物的构效关系(structure-activity relationship,SAR)进行研究后首次制备了苯并噻唑15,该化合物可产生S1PR1激动作用(EC50=0.041 μmol·L-1),且对S1PR3有选择性(EC50=1.21 μmol·L-1),但其较低的总极性表面积(tPSA=52.9 Å2)和相对较高的亲脂性(clogP=3.92)降低了安全性[45]。为了提供有效、选择性,且具有较高体内淋巴细胞增殖活性、较低亲脂性和较高总极性表面积的S1PR1激动剂,Frohn 等[45]在化合物15 的基础上进行结构改造,在其“核心”区域引入极性,最终得到S1PR1激动剂AMG 369(16,EC50=0.002 μmol·L-1),该化合物对S1PR3有选择性(EC50=0.888 μmol·L-1)。
随后,Frohn 等[45]在AMG 369 基础上继续进行了结构修饰:以氮(杂)环丁烷-3-羧酸为极性头,苯并噻唑为连接臂,主要考察增加疏水侧链部分的极性对化合物活性和选择性的影响。结果发现,引入疏水侧链甲酰胺基对化合物的活性和选择性有很大的提升。所得化合物中,(+)-17 的活性最好,且对S1PR1的选择性是对S1PR3的23 倍(EC50分别为0.017 和0.39 μmol·L-1)。由此证明以苯并噻唑类的稠环作为连接臂是可行的,且极性较大的甲酰胺的疏水侧链打破了大多数的S1PR1选择性激动剂的疏水侧链都是烷基或烷氧基的常规模式,这一结构修饰具有非常大的创新性。
2.2.3 塞拉利莫德Kurata 等[46]通过筛选他们的化合物库最终得到了亲水性头部区域含有β-丙氨酸部分,亲脂性尾部区域含有5-苯基戊基部分的化合物18。在钙离子内流试验中,化合物18 对S1PR1的选择性是对S1PR3的26 倍,在小鼠外周淋巴细胞降低(peripheral lymphocyte lowering,PLL)试验中,ED50为26 mg·kg-1(经口给药后4 h)。随后,在化合物18 的中心区域和亲水性氨基酸区域引入双键和闭合环来限制构象,最终得到具有中心二氢萘核的选择性的先导化合物19。化合物19 对S1PR1的选择性是对S1PR3的26 000 倍,且在小鼠PLL试验中有良好疗效(ED50=0.22 mg·kg-1,经口给药后24 h)。随后,在化合物19 基础上进一步优化二氢萘系列中的亲脂性尾部区域,从而发现了比化合物19 更有效的临床候选药物塞拉利莫德(20)。化合物20 是选择性、高效的S1PR1和S1PR5激动剂(EC50分别为27.3 和334 pmol·L-1)。该化合物在钙离子内流试验中对S1PR1的选择性是对S1PR3的30 000 倍,在小鼠PLL 试验中非常有效(ED50=0.029 mg·kg-1,经口给药后24 h)。塞拉利莫德治疗RRMS 患者(临床试验编号:NCT01226745)的Ⅱ期临床扩展试验已完成,研发公司已决定不再进行塞拉利莫德的第3 阶段开发,该决定与任何安全性和有效性调查结果无关。

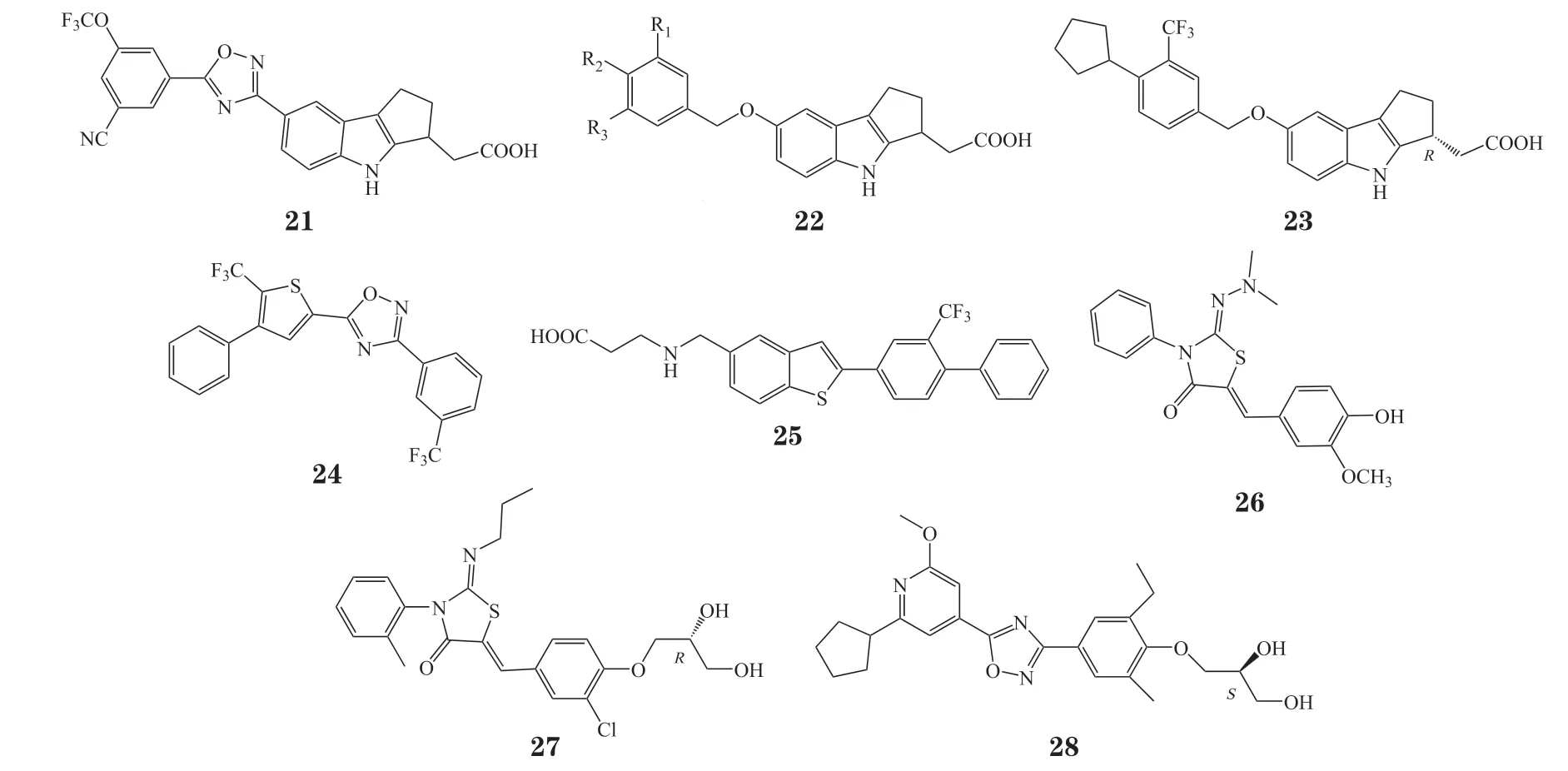
2.2.4 艾曲莫德Buzard 等[47]早期着重优化5-苯基-1,2,4-二唑类化合物(如化合物21),尽管发现了几种有效且有选择性的类似物,但在小鼠PLL 试验中,该类化合物未能显示良好活性。此外,静脉注射24 h 后,在大鼠大脑和脑脊液中检测不到化合物21,然而中枢暴露被认为是成功治疗MS 的必要条件[48]。为了提高化合物的中枢暴露,Buzard 等[49]将连接臂二唑替换为苄氧基(22),发现R1和R2双取代的化合物活性更好,最终得到了艾曲莫德(23),对S1PR1表现较强激动作用(EC50=6.10 nmol·L-1),对S1PR4和S1PR5表现出较弱激动作用(EC50分别为147 和24.4 nmol·L-1)。2022 年3 月24 日辉瑞(Pfizer)公布了艾曲莫德一项Ⅲ期临床研究ELEVATE 12(临床试验编号:NCT03996369)的阳性顶线结果,数据显示艾曲莫德在治疗中度至重度活动性溃疡性结肠炎(ulcerative colitis,UC)中有显著疗效。
2.2.5 SEW2871 和AUY954前期研究中,通常将S1PR1激动剂的结构分为极性头、连接臂和疏水侧链3 部分。通过高通量药物筛选发现的SEW2871(24)及其衍生物是第1 类没有极性头部的选择性S1PR1激动剂[50],SEW2871 对S1PR1的 EC50为0.27 μmol·L-1,而且对 S1PR3的选择性也较好[51]。科学家对这类化合物与S1PR1受体的相互作用方式进行了分子模拟,认为SEW2871 结构中的三氟甲基相当于极性头部,分子中的三环刚性侧链是这类化合物具有选择性的原因。然而,作为早期发现的先导化合物,其活性与芬戈莫德相比不甚理想[52]。
AUY954(25)的研发灵感来源于将高通量药物筛选中获得的线索和与SEW2871 相关的筛选线索结合后所得出的结构特征。AUY954 是将不同的氨基羧酸“头基”安装到具有三氟甲基联苯药效团的各种苯并稠杂环上获得的有效且选择性好的S1PR1激 动剂(EC50=1.2 nmol·L-1),对 S1PR1的亲和力是其他受体亚型的280 倍。AUY954 在大鼠体内可引起长时间的、可逆的外周淋巴细胞减少,且具有良好的药代动力学性质[53]。目前,SEW2871和AUY954 均未进入临床。
2.3 以甘油为头部的化合物
2.3.1 波西莫德Bolli 等[54]通过高通量筛选得到具有S1PR1激动作用的含2-亚氨基-4-噻唑烷酮骨架的化合物26,该化合物对S1PR1和S1PR3的EC50分别为200 nmol·L-1和1 μmol·L-1。进一步以化合物26 为先导物进行结构改造,发现以R-甘油为头部的波西莫德(27)无论在与33P-S1PR 的结合试验(IC50=6.0 nmol·L-1)中,还是在GTPγS 结合试验(EC50=5.7 nmol·L-1)中,都表现出对S1PR1较高的选择性。波西莫德的心脏安全性良好,在优化治疗方案后,该药可以最大限度地减少与S1PR1对心肌细胞的调节相关的首剂效应[55]。目前已完成波西莫德在有症状的慢性GVHD 受试者中的生物活性、安全性、耐受性和药代动力学的Ⅱ期临床研究(临床试验编号:NCT02461134);正在进行波西莫德(20 mg)在复发型MS 患者中的安全性、耐受性和疾病控制的Ⅲ期临床试验(临床试验编号:NCT03232073)。
2.3.2 西奈莫德Bolli 等[56]创新性地提出以3-苯基-5-(吡啶-4-基)-1,2,4-二唑为化合物骨架进行结构改造,这类结构具有强大且持久的免疫调节作用,最终得到高效的选择性S1PR1激动剂西奈莫德(28,EC50=1 nmol·L-1)。西奈莫德在表达重组S1PR1的细胞和非重组人内皮细胞中显示出明显的S1PR1介导的通路偏爱性,导致细胞内钙离子动员反应显著减弱。芬戈莫德、阿米莫德、辛波莫德和天然配体S1P 均未显示出这种通路偏爱性,这表明西奈莫德显示出独特的S1PR1信号特性。与芬戈莫德不同,西奈莫德在体内和体外均没有血管收缩和支气管收缩的潜力,这可能是由于S1PR1选择性和S1PR1信号传导偏爱性的独特组合[57]。目前,正在进行西奈莫德的多项临床试验,其中一项Ⅱ期临床试验(临床试验编号:NCT03742037)考察了4 种剂量的西奈莫德对活动性系统性红斑狼疮成年受试者的疗效和安全性。
3 总结与展望
S1P 及其受体参与许多生理和病理过程,通过调节免疫细胞迁移、驱动免疫细胞分化、改变其功能表型,在免疫应答中发挥关键作用。目前,有临床前证据支持S1PR 调节剂在重症肌无力、帕金森病、阿尔茨海默病、亨廷顿病、胃癌、1 型糖尿病、自身免疫性心肌炎和扩张型心肌病、肥厚型心肌病、败血症等疾病中存在潜在治疗作用[58]。此外,有证据显示对S1P 合成、降解和转运途径的调节也可能代表了新的药物靶点。例如,在IBD 患者的肠道中观察到S1P 通路的失调;SphK1 上调与结肠炎相关性癌症和其他癌症(弥漫性B 细胞淋巴瘤、乳腺癌、结直肠癌、食管癌、胃癌和前列腺癌)有关[59]。这些临床前研究结果为进一步研究S1PR 调节剂及发现新的治疗靶点提供了证据支持。
目前,对于S1PR1调节剂的研发依旧主要是提高S1PR1调节剂的选择性及改善药代动力学性质,通过结构修饰和改造得到选择性更好、副作用更小的S1PR1选择性调节剂。本文列举了不同类别的S1PR1选择性调节剂,包括已经上市的药物和正在进行临床研究的药物,以期对S1PR1选择性调节剂的研究提供大致的思路,对进一步研究S1PR1调节剂提供参考价值。
