靶向核受体的抗前列腺癌小分子药物研发新进展
2023-10-20袁乐儿廖金标蔡吕涛胡陈娴杨号东盛荣
袁乐儿,廖金标,蔡吕涛 ,胡陈娴,杨号东,盛荣*
(1.浙江大学药学院,浙江 杭州 310058;2.浙江大学金华研究院,浙江 金华 321002)
2022 年全球癌症统计资料显示,前列腺癌(prostate cancer,PCa)已成为男性发病率第2 的癌症,致死率排名第5[1]。前列腺癌发病机制复杂,影响因素众多。研究表明,核受体家族蛋白在前列腺癌的发生发展中具有重要作用,核受体主要有雄激素受体(androgen receptor,AR)、糖皮质激素受体(glucocorticoid receptor,GR)和盐皮质激素受体(mineralocorticoid receptor,MR)等。其中,AR 信号通路在前列腺癌发生和发展过程中起关键作用[2],拮抗AR 通路是治疗激素敏感性前列腺癌及去势抵抗性前列腺癌(castration-resistant prostate cancer,CRPC)的重要策略。目前,第2 代AR 拮抗剂恩杂鲁胺已成为一线治疗药物,但随着AR 拮抗剂的广泛使用,临床耐药已经产生,耐药机制包括AR基因扩增、AR 配体结合域上的点突变和缺乏配体结合域(ligand-binding domain,LBD)的剪接变体(AR variants,ARVs)产生[3]。
临床研究发现,CRPC 的耐药往往伴随着GR 表达的上调。由于AR 和GR 高度相似,在CRPC 中,GR可绕过AR 阻断,代替AR 调控AR 蛋白的表达,促进肿瘤细胞增殖[4]。研究表明,GR 拮抗剂可恢复前列腺癌细胞对AR 拮抗剂的敏感性,因此,同时阻断AR 和GR 信号通路成为克服CRPC 耐药性的有效策略[5]。
本文对近5 年(2018—2023)靶向核受体AR和GR 的抗前列腺癌药物研发新进展进行综述,包括雄激素竞争性AR 拮抗剂、雄激素非竞争性AR拮抗剂、AR 降解剂、GR 拮抗剂和AR/GR 双重拮抗剂,旨在为CRPC 的治疗提供新思路。
1 雄激素受体
1.1 雄激素受体的结构及功能
AR 是由919 个氨基酸组成的类固醇受体转录因子,为核受体家族成员。AR基因位于染色体Xq12上,由8 个外显子组成,如图1A 所示。AR 由N 端结构域(N-terminal domain,NTD)、DNA 结合域(DNA-binding domain,DBD)、铰链区(hinge,H)和LBD 组成。外显子1 编码的NTD 包含激活功能区1(activation function 1,AF1),支持AR 的转录活性。外显子2 和3 编码的DBD 包含2 个锌指结构,第1 个锌指结构决定DNA 结合特异性,第2个锌指结构与AR 二聚化及DNA 受体复合物的稳定性有关。C 端包含由外显子4 编码的柔性铰链区和外显子5~ 8 编码的高度保守的LBD,LBD 包含配体结合口袋(ligand-binding pocket,LBP)、激活功能区2(activation function 2,AF2)和结合功能区3(binding function 3,BF3)[6]。
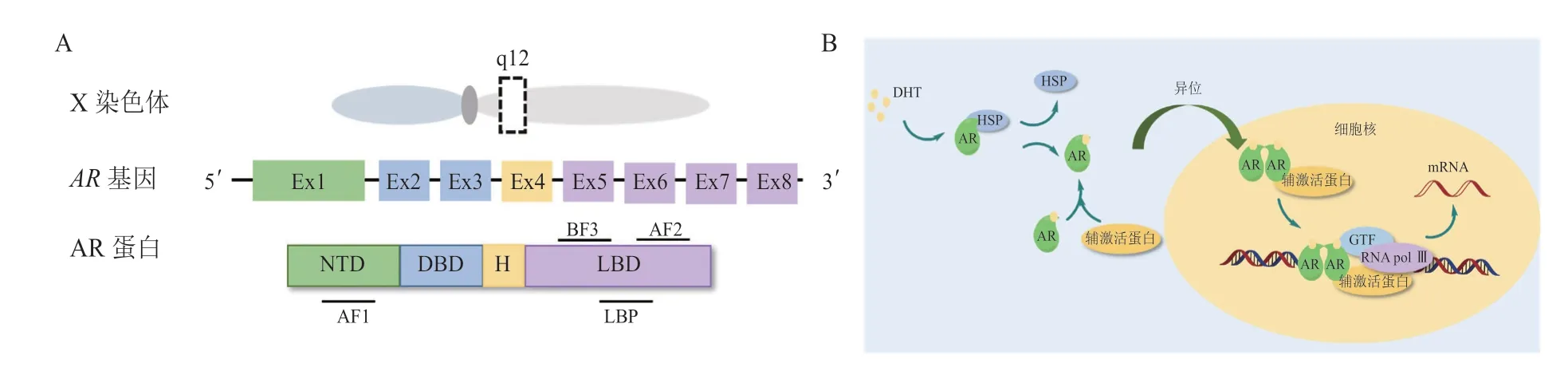
图1 雄激素受体的结构及其信号通路Figure 1 Structure of androgen receptor and its signaling pathway
1.2 雄激素受体的信号通路
AR 的信号通路如图1B 所示,未与雄激素[睾酮或二氢睾酮(dihydrotestosterone,DHT)]结合的AR 蛋白位于细胞质中,与热休克蛋白(heat shock protein,HSP)形成稳定的复合物。雄激素与AR 的LBD 中的LBP 结合后,诱导AR 构象发生变化,随后,AR 与HSP 分离,发生同源二聚化,并易位至细胞核内,二聚化AR 的DBD 与DNA 上的雄激素应答元件(androgen response element,ARE)结合后,招募一系列转录共调节因子,进而调节近百种AR 靶基因的表达,包括前列腺特异性抗原(prostate-specific antigen,PSA)、跨膜丝氨酸蛋白酶2(transmembrane protease serine 2,TMPRSS2)等,该过程的过度激活促进前列腺癌的进展[7]。
2 雄激素竞争性雄激素受体拮抗剂
AR 的激活高度依赖于雄激素与LBP 的结合,因此靶向AR 的LBP 拮抗雄激素的作用,是近年AR 拮抗剂新药研发的主要策略。目前已上市的第1代和第2 代AR 拮抗剂(结构式见图2)均结合于AR LBP 位点,其作用机制为LBP 与雄激素竞争性结合从而阻断AR 激活,进而抑制AR 信号通路,降低血清PSA 水平[6]。
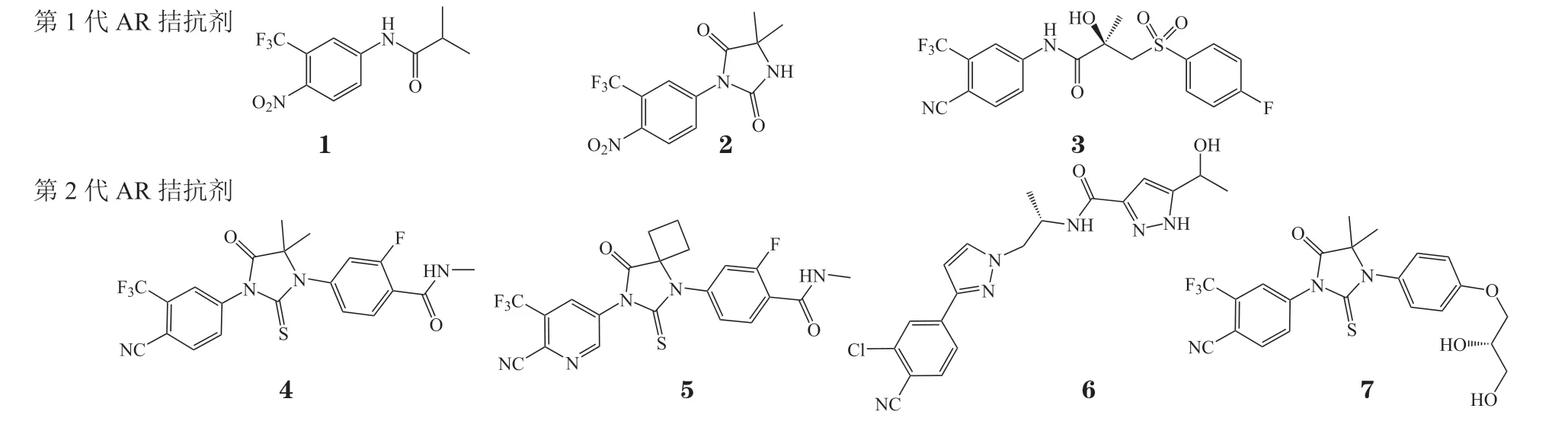
图2 第1 代和第2 代雄激素受体竞争性拮抗剂的化学结构式Figure 2 Chemical structures of the first and second generations of androgen receptor competitive antagonists
2.1 第1 代竞争性雄激素受体拮抗剂
氟他胺(flutamide,1)于1989 年获美国食品药品监督管理局(Food and Drug Administration,FDA)批准用于治疗PCa,是首个上市非甾体类AR 拮抗剂。通过进一步的结构优化得到的尼鲁他胺(nilutamide,2)和比卡鲁胺(bicalutamide,3),分别于1995 年 和1996 年获批。第1 代AR 拮抗剂与AR 的亲和力较弱,不能充分阻断AR 通路,与雄激素剥夺疗法(androgen-deprivation therapy,ADT)联用可适度延长患者总生存期,但不良反应发生率较高,且长期用药易引起AR LBD 的耐药突变,包括AR W742C/L 和H875Y/T878A 突变等[6],导致拮抗剂转变为部分激动剂,使患者产生耐药性,最终发展为CRPC[8]。
2.2 第2 代竞争性雄激素受体拮抗剂
恩杂鲁胺(enzalutamide,4)是首个第2 代AR拮抗剂,与AR 结合亲和力显著提升,能抑制AR核易位、与雄激素结合以及与靶基因结合的全过程,分别于2012 年、2018 年和2019 年被FDA 批准用于治疗转移性去势抵抗性前列腺癌(metastatic castration-resistant prostate cancer,mCRPC)、非转移性去势抵抗性前列腺癌(non-metastatic castrationresistant prostate cancer,nmCRPC)和激素敏感性前列腺癌(hormone-sensitive prostate cancer,HSPC)[9];但是因在患者脑内恩杂鲁胺稳态水平较高,导致拮抗γ-氨基丁酸α(γ-aminobutyric acid α,GABAα)受体从而易产生癫痫等不良反应。阿帕鲁胺(apalutamide,5)为恩杂鲁胺的类似物,该药对AR 的拮抗活性更高,分别于2018 年和2019 年获批用于治疗nmCRPC 和mHSPC[9],且不易入脑,中枢神经系统(central nervous system,CNS)不良反应较少。但AR LBD 的F877L 突变使恩杂鲁胺和阿帕鲁胺转变为激动剂,以及ARVs的产生也导致耐药[10]。
达洛鲁胺(darolutamide,6)由拜耳和奥利安公司联合研发,该药能有效抑制恩杂鲁胺和阿帕鲁胺耐药的AR F877L 突变体,延长高危nmCRPC 患者的无转移生存期,于2019 年获FDA 批准用于治疗nmCRPC[11]。但是患者发生疲劳、疼痛等副作用的比例较高,导致临床应用受限[12]。
恒瑞医药研发的瑞维鲁胺(rezvilutamide,7)也为恩杂鲁胺类似物,在mCRPC 的Ⅰ/Ⅱ期临床试验中,该药160 mg·d-1剂量与360 mg·d-1恩杂鲁胺的药物暴露量相当,且耐受性良好[最大耐受剂量(maximal tolerance dose,MTD)大于480 mg·d-1];同时,该药还具有血脑屏障透过率低、诱发癫痫风险小和安全性高等优点[13]。瑞维鲁胺联合ADT 治疗高瘤负荷mHSPC,可显著延长患者总生存期,于2022 年6 月获国家药品监督管理局(National Medical Products Administration,NMPA)批准治疗mHSPC[14]。
目前,还有多种AR 拮抗剂处于临床试验阶段(结构式见图3)。例如,HC-1119(8)为氘代恩杂鲁胺,针对mCRPC 患者的Ⅰ期临床试验显示,与恩杂鲁胺相比,HC-1119 代谢缓慢、半衰期更长,母体药物的血浆浓度增加约40%,主要代谢物的血浆浓度降低约75%[15],目前正开展治疗mCRPC 上市申请。
开拓药业的普克鲁胺(9)与AR 的结合亲和力比恩杂鲁胺高3.4 倍[半数抑制浓度(half maximal inhibitory concentration,IC50)为32 nmol·L-1,抑制常 数(inhibition constant,Ki)为14 nmol·L-1)],且对ARF877L,ARW742C/L和ARH875Y/T878A等耐药突变体有效。该药在体内消除速度慢,单次和连续给药的平均表观清除率分别为0.55~ 1.30 和0.17~ 0.46 L·h-1[16],且CNS 分布低,不易诱发癫痫,mCRPC 的Ⅱ期临床试验效果良好[17]。
西安杨森的TRC-253(10)可强效抑制野生型AR 和F877L 突变体,IC50分别为54 和37 nmol·L-1,在F877L突变体的异种移植模型中抑瘤效果显著[18],正处于mCRPC Ⅱ期临床[19]。
目前,正大天晴的TQB3720[20]、日本大鹏药品的TAS-3681[21]、康朴的X-Synergy®(结构均尚未披露)处于Ⅰ期临床研究。
此外,还有多种AR 拮抗剂正处于临床前研究阶段(结构式见图3)。其中,化合物11 在LNCaP细胞中的AR 转录抑制活性与恩杂鲁胺相近[8],化合物12 在LNCaP 细胞中的活性较恩杂鲁胺提高近60 倍[22]。(+)JJ-450(13)经高通量筛选和结构优化得到[23],在C4-2-PSA-rl 细胞中,其活性与恩杂鲁胺相当,作用机制为延缓AR 核易位,且对恩杂鲁胺耐药的CRPC 有效,能抑制剪接变体AR-V7 的转录活性和基因表达[24]。苯甲酰衍生物14 和15,对表达AR野生型(wild-type,WT)、T877A 和H874Y 突变体的前列腺癌细胞,均具有显著的增殖抑制作用[25]。
侯廷军团队基于AR LBD 结构,经虚拟筛选发现AT2(16)为全新骨架AR 拮抗剂,其抗AR 转录活性的IC50为0.06 µmol·L-1,能抑制AR 下游靶基因及AR 核易位[26]。该团队采用分子动力学模拟构造AR LBD 二聚体结构,并进行虚拟筛选和结构优化,其中香豆素类衍生物17 抑制AR 转录的IC50为0.17 µmol·L-1,机制可能是抑制AR 二聚化[27]。
以达洛鲁胺为先导化合物,对其2-氯苯腈部分和吡唑部分进行结构改造,其中,类似物18 能有效拮抗AR 转录活性(IC50=0.05 µmol·L-1),对ARF876L和ART877A的效力均优于达洛鲁胺,且抑制AR 下游靶基因表达[28]。
3 非雄激素竞争性雄激素受体拮抗剂
AR LBP 易突变的性质严重限制经典AR 竞争性拮抗剂的临床使用。近年来,靶向AR 雄激素非竞争性位点的拮抗剂也得到了发展,如靶向LBD 的AF2 和BF3,NTD 的AF1 以及DBD 的拮抗剂(结构式见图4),以寻求克服耐药的前列腺癌新疗法。

图4 雄激素受体非雄激素竞争性拮抗剂的化学结构式Figure 4 Chemical structures of androgen receptor nonandrogen competitive antagonists
3.1 靶向配体结合域激活功能区2 的拮抗剂
AF2 是AR 与雄激素结合发生构象变化后在LBD 表面形成的疏水口袋,在该位点招募共调节因子对AR 发挥转录活性至关重要。AF2 可通过与共调节因子LXXLL 和FXXLF 基序特异性相互作用来募集辅助激活因子。AF2 作为转录激活结构域,在正常细胞中调节功能较弱,而在CRPC AR 高表达的环境中趋于主导。因此,靶向AF2 直接阻断AR 与辅助激活因子相互作用的疗法,理论上不会受LBP耐药突变的影响,从而具有良好的临床应用前景[29]。
双苯甲酰胺类衍生物20 的AR 转录抑制活性优异,IC50为16 nmol·L-1,且100 nmol·L-1浓度下能有效抑制LNCaP 细胞活性,并抑制AR 调控基因PSA的表达。机制研究表明,该化合物在AF2 位点抑制AR 与共调节因子PELP1 的相互作用,从而发挥AR 拮抗活性[31]。
基于AF2 蛋白进行虚拟筛选发现的磺酰胺类化合物T1-12(21),其AR 转录抑制活性优良,IC50为 0.47 μmol·L-1,能显著抑制AR 下游基因表达和AR 核易位,在LNCaP 异种移植模型的效果与恩杂鲁胺相当。经时间分辨荧光共振能量转移技术(timeresolved fluorescence resonance energy transfer,TRFRET)AR 共激活因子试剂盒验证,T1-12 能浓度依赖性地下调FRET 信号,通过阻断AF2 与共调节因子FXXLF 结合,从而发挥AR 拮抗活性[32]。
3.2 靶向配体结合域结合功能区3 的拮抗剂
BF3 位点在空间上与AF2 相邻,BF3 通过招募FK506 结合蛋白52(FK506 binding protein 52,FKBP52)和Bcl-2 结合抗凋亡蛋白1(Bcl-2-associated athanogene-1,BAG-1)等辅助激活因子来调控AR转录活性。同时,BF3 还与相邻的AF2 形成变构网络以调节AR LBD 功能。前列腺癌和雄激素不敏感综合征中大量的BF3 突变(前者如Gln670 等;后者如Leu830,Pro723 等),以及BF3 抑制剂氟灭酸的发现,证明靶向BF3 作为治疗前列腺癌策略的可行性[33]。
Leblanc 等[34]发现2-(1H-吲哚-3-基)喹啉衍生物VPC-13566(22)具有良好的AR 转录抑制活性(IC50=0.06 μmol·L-1),但体内代谢不稳定,半衰期短(T1/2=21 min)。对吲哚及喹啉环进行修饰后得到的衍生物VPC-13789(23),代谢稳定性显著提高(T1/2=206 min),能有效抑制WT-AR(IC50=0.19 μmol·L-1)和ARF877L突变体(IC50=0.21 μmol·L-1)的活性,抑制LNCaP 细胞的增殖。将VPC-13789 制备为可口服的磷酸钠前药VPC-13822(24),VPC-13822 在CRPC 异种移植模型中疗效良好,显著降低血清PSA 水平,且长期毒性小,具有明显的临床价值。
Chen 等[35]基于恩杂鲁胺和达洛鲁胺化学结构,经骨架跃迁设计得到一系列衍生物,其中化合物25的AR 转录抑制活性优良(IC50=0.07 μmol·L-1),对ARF877L/T878A双突变体有效(IC50=0.25 μmol·L-1),且能有效抑制LNCaP 细胞增殖(IC50=6.23 μmol·L-1),在小鼠LNCaP 异种移植瘤模型中,经口给药(100 mg·kg-1·d-1)28 天后,可有效抑制肿瘤生长,分子动力学模拟结果预测该化合物可能结合于BF3位点。
3.3 靶向配体结合域二聚体界面口袋的拮抗剂
侯廷军团队通过分子动力学模拟和小角度X 射线散射实验,发现AR 的二聚体界面口袋(dimer interface pocket,DIP),并通过基于结构的虚拟筛选,发现小分子拮抗剂M17-B15(26)能够有效地破坏AR 蛋白二聚化,进而抑制AR 信号转导。其AR 转录抑制活性优良(IC50=0.03 μmol·L-1),且对恩杂鲁胺耐药的ARF876L/T877A双突变体有效(IC50=0.15 μmol·L-1),能有效抑制AR 调控及基因转录和翻译水平。在异种移植LNCaP 细胞模型中,瘤内注射M17-B15(2.5 mg·kg-1·d-1)可显著抑制肿瘤生长。因此,靶向DIP 位点是全新的AR 拮抗剂研发策略[36]。
是被陕西来的一个戏班子启封的。说来这似乎应当和那戏班子里一名女子有那么一点关系。那个女子也胖大(同他这次找来的那个胖大的女子竟有些相像,有命里注定却已然隔世的意思),秦腔的唱音既宏阔还自带扩音效果,能将那木偶像他现在这样耍得活灵活现跟个真人儿似的。甚至,她连在台下的一举手一投足都满满地带着一股子戏派,简直迷住了其时四十多岁的王爷。
3.4 靶向N 端结构域的拮抗剂
NTD 是AR 完全转录活性的关键区域,并存在于所有形式的AR 中,且NTD 可以在雄激素非依赖性前列腺癌细胞中调节AR 活性[21]。因此,靶向NTD 的拮抗剂有望解决CRPC 耐药性问题。
从海绵提取物库中筛选而得的EPI-001 的最有效的立体异构体为EPI-002(27),该化合物能与NTD 上的TAU-5 结合[37],其乙酰基前药 EPI-506(28)为首个进入临床的NTD 拮抗剂,但因药物剂量负担过重、口服生物利用度差而终止试验[38]。类似物EPI-7386(29)在LNCaP 异种移植模型中活性与恩杂鲁胺相当;在恩杂鲁胺耐药的VCaP 异种移植模型中,EPI-7386 单药或与恩杂鲁胺联用,均表现出显著的抗肿瘤活性,目前处于mCRPC 的Ⅰ/Ⅱ期临床试验[39]。
通过靶向AR NTD 的虚拟筛选获得的QW07(30),具有良好的AR 转录抑制活性(IC50=4.93 μmol·L-1),能有效抑制LNCaP 和22RV1 细胞增殖。在CRPC 动物模型中,QW07(10 mg·kg-1·d-1)显著抑制22RV1 和VCaP 肿瘤的生长。染色质免疫沉淀实验证明,QW07 与AR NTD 的结合抑制AR 转录复合物的形成,从而阻止下游基因与启动子、增强子的结合[40]。
研究发现DHT 可诱导AR 入核,并发生液-液相分离,从而形成激活的转录凝集体,该过程主要由NTD 驱动,采用ARF877L/T878A细胞株对化合物库进行筛选,获得化合物ET0516(31),并经微尺度热电泳等实验证明该化合物结合在AR NTD。ET0516 可有效地抑制野生型和耐药突变的AR 的相分离形成,在5.0 μmol·L-1浓度下,对LNCaP 和VCaP 细胞增殖抑制率大于50%[41]。
3.5 靶向DNA 结合域的拮抗剂
DBD 是AR 与AREs 结合不可或缺的结构域,对AR-FL 和雄激素非依赖ARVs 的核定位至关重要。因此,靶向DBD 的拮抗剂可以直接阻断 AR 与DNA 的相互作用,以克服CRPC 耐药性[42]。
基于DBD 结构虚拟筛选发现的噻唑吗啉衍生物VPC-14449(32)具有优良的AR 转录抑制活性(IC50=0.34 μmol·L-1),能有效抑制LNCaP 细胞和恩杂鲁胺耐药MR49F 细胞的增殖,生物膜干涉实验结果证明,该化合物选择性地靶向AR DBD-铰链区结构域表面,进而阻断AR 与DNA 相互作用[43]。
二氢查尔酮衍生物MF-15(33)为AR 和AKR1C3 的双重抑制剂。AKR1C3 为参与雄激素生物合成的酶,与恩杂鲁胺耐药CRPC 有关。在10 µmol·L-1浓度下,MF-15 对AKR1C3 的抑制率为87%,并能浓度依赖性地抑制AR-FL和AR-V7的活性,显著抑制AR 下游PSA表达。此外,MF-15 抑制DBD中的P-box 相互作用而发挥AR 拮抗作用[44]。
基于DBD 晶体的虚拟筛选发现了苯甲酸衍生物Cpd39(34),其AR 转录抑制活性中等(IC50=10.94 μmol·L-1),能减少WT-AR 和AR-V7 调控基因的表达,且抑制AR 下游PSA基因表达,生物膜干涉实验结果表明,该化合物靶向DBD-ARE 结合界面位点,抑制AR DBD-DNA 相互作用[45]。
4 雄激素受体降解剂
4.1 选择性雄激素受体降解剂
临床患者对AR 拮抗剂耐药的快速出现,使得对靶向AR 的新策略需求增加,其中选择性雄激素受体降解剂(selective androgen receptor degrader,SARD)是克服耐药的策略之一(结构式见图5)[46]。目前,SARD 的确切作用机制尚不明确,一般认为是通过泛素-蛋白酶体系统(ubiquitin-proteasome system,UPS)介导AR 的降解,可能是通过增强AR 与E3连接酶——双微体同源基因2(murine double mimute 2,MDM2)的关联而发挥作用。以靶向LBP 的AR拮抗剂RU59063 母核作为AR 配体,并通过烷基链和三氮唑连接疏水标签(hydrophobic tag,HyT),得到化合物A9(35),其AR 转录抑制活性良好(IC50=1.75 μmol·L-1),使 用10 μmol·L-1A9 处 理LNCaP 细胞72 h 后,可基本实现AR 的完全降解[47]。
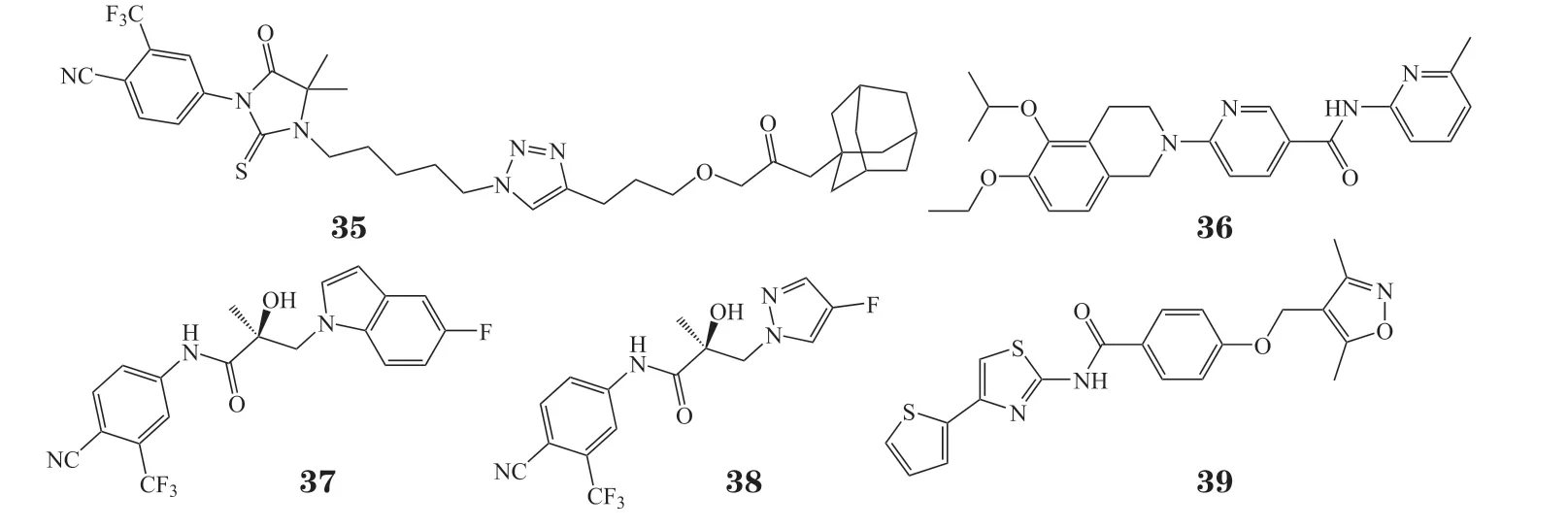
图5 选择性雄激素受体降解剂的化学结构式Figure 5 Chemical structures of selective androgen receptor degrader
经虚拟筛选发现的二氢异喹啉烟酰胺衍生物EIQPN(36),其作用位点为AF1,在10 μmol·L-1浓度下,其AR 抑制率大于95%,对LNCaP 细胞增殖抑制率大于90%。在NTD 过表达HEK293T 细胞中,EIQPN 的IC50为0.78 μmol·L-1。作用机制研究表明,EIQPN 能有效降低LNCaP,CWR22rv,DU145,PPC1 和HEK293T 细胞的AR 和ARVs 水平,且能抑制异种移植小鼠模型CWR22rv 肿瘤生长[48]。
对AR 拮抗剂比卡鲁胺采用合环策略,得到类似物UT-155(37)和UT-34(38)。作用机制研究表明,两者均能与AF1 位点结合,并具有SARDs功能。体外活性测试表明,在1.0 μmol·L-1浓度下,两者在LNCaP 和22RV1 细胞系中均能有效降解全长AR 和ARVs,并对恩杂鲁胺耐药MR49F 细胞生长抑制率大于70%。在100 mg·kg-1剂量下,UT-155 和UT-34 在LNCaP 去势模型和MR49F 模型中均能有效抑制肿瘤生长[49]。
Wu 等[50]经药效团虚拟筛选发现了小分子SARD Z15(39),靶点验证发现该化合物为AF1 与LBD双位点SARD。体外活性测试表明,Z15 在LNCaP细胞中可明显下调AR 蛋白表达水平,半数最大降解浓度(the half-maximal degradation concentration,DC50)为1.05 μmol·L-1。5 μmol·L-1的Z15 和环己酰胺(100 μg·mL-1)联用处理LNCaP 细胞24 h 后,可基本实现AR 完全降解。在22Rv1 细胞中,Z15 对AR 和AR-V7 的DC50分别为1.16 和2.24 μmol·L-1。
4.2 雄激素受体蛋白降解靶向嵌合体
作为一种新兴的靶蛋白降解技术,蛋白降解靶向嵌合体(proteolysis targeting chimera,PROTAC)近年来迅速发展,引起越来越多的关注。PROTAC由3 个部分组成:与E3 泛素连接酶结合的配体,与靶蛋白(protein of interst,POI)结合的配体以及连接2 个配体的连接链。PROTAC 分子与E3 泛素连接酶和POI 形成三元复合物,特异性诱导POI 的泛素化标记,进而通过泛素-蛋白酶体途径降解多泛素化的靶蛋白[51]。
第1 个靶向AR 的PROTAC 分子是含有E3 泛素连接酶的肽基配体,其理化性质和细胞通透性较差,限制了进一步的应用。2008 年,Crews 等[52]首次报道以MDM2 抑制剂为E3 连接酶配体,比卡鲁安作为AR 配体的小分子AR PROTAC,该化合物能在微摩尔浓度下降解AR 蛋白,从而开启小分子AR PROTAC 药物(结构式见图6)时代。
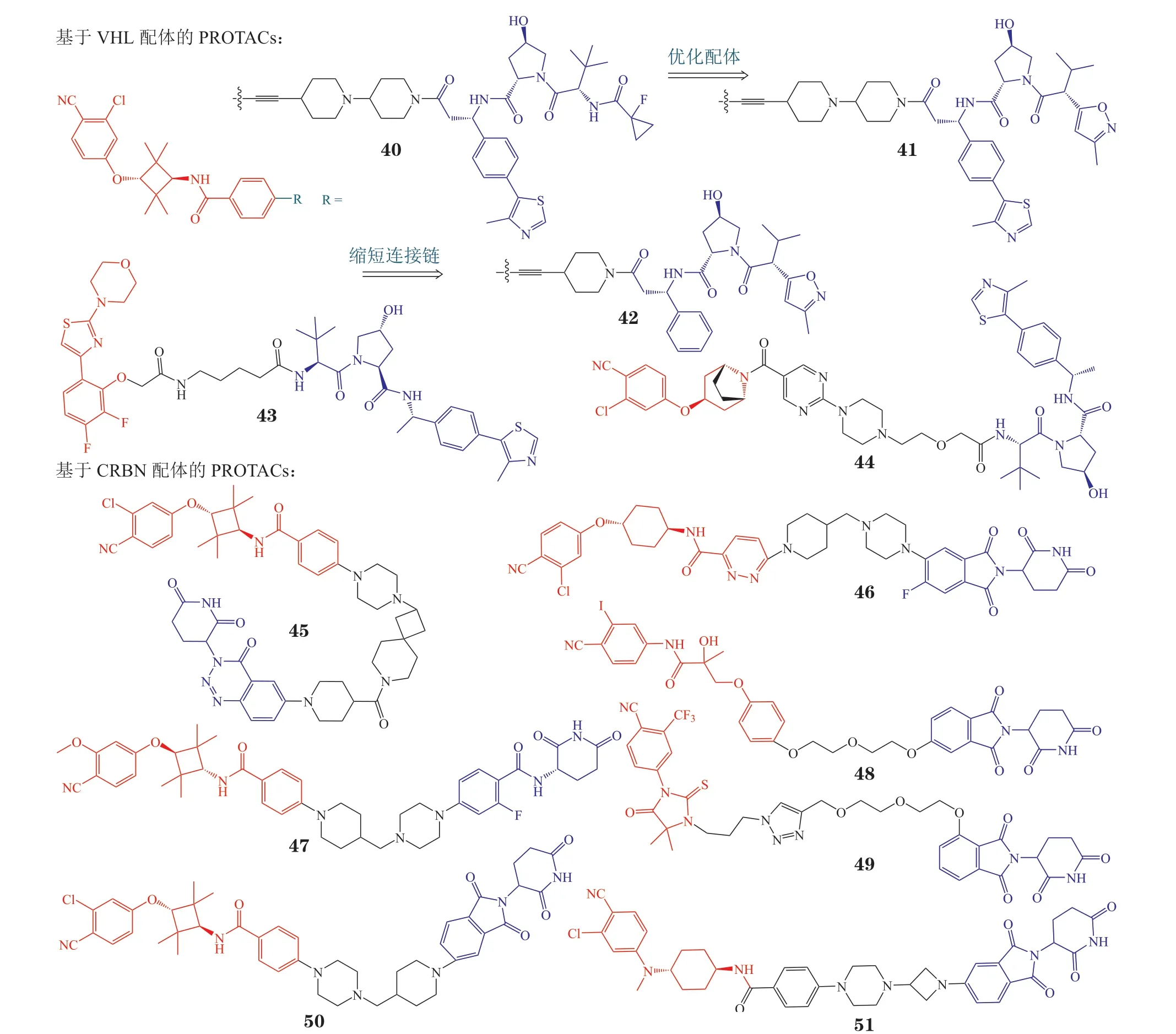
图6 雄激素受体蛋白降解靶向嵌合体的化学结构式Figure 6 Chemical structures of androgen receptor proteolysis targeting chimera
4.2.1 基于希佩尔-林道E3 泛素连接酶配体的蛋白降解靶向嵌合体芳氧基环丁基胺衍生物是高活性AR拮抗剂[53],其骨架常作为AR 配体,以希佩尔-林道(von Hippel-Lindau,VHL)配体为E3 泛素连接酶配体,通过刚性接头将两者连接得到PROTAC 分子ARD-69(40)[54]。对ARD-69 的活性进行评估,实验结果表明,ARD-69 在LNCaP,VCaP 和22Rv1细胞中的DC50分别为0.86,0.76 和10.4 nmol·L-1。细胞增殖抑制实验中,该化合物对LNCaP,VCaP 和22Rv1 细胞的IC50分别为0.25,0.34 和183 nmol·L-1,活性为恩杂鲁胺的100 倍以上。腹腔给药50 mg·kg-1ARD-69 后,在48 h 内有效减少VCaP 异种移植小鼠的AR 蛋白和PSA 蛋白。ARD-61(41)是优化VHL 配体得到的新PROTAC 分子[55],表现出良好的体内外AR 降解活性,且对恩杂鲁胺耐药模型以及AR 阳性乳腺癌有效。
对ARD-61 进一步进行结构修饰,将不同AR配体与VHL 配体进行组合得到ARD-266(42)[56]。ARD-266 在1 nmol·L-1浓度下 即可诱 导LNCaP,VCaP 和22Rv1 细胞的AR 蛋白降解,与ARD-61相比,ARD-266 在保持良好的AR 降解效率和前列腺癌拮抗活性的同时,具有更小的相对分子质量,且理化性质和成药性更佳。
MTX-23(43)由DBD 配体、柔性连接链和VHL 配体组成[57],由于该化合物结合于DBD,因此对AR-FL 和AR-V7 具有显著降解活性,DC50分别为2.00 和0.37 µmol·L-1,它能有效抑制CRPC 细胞增殖,经口给药(8.3 mg·kg-1)显著抑制恩杂鲁胺耐药的前列腺癌异种移植瘤(如22Rv1 肿瘤),且MTX-23 对AR-V7 的降解效果优于AR-FL,其深入的机制仍在探索中。
化合物A031(44)由具有芳氧基托品环的独特AR 配体、VHL 配体和包含哌嗪和嘧啶环的连接体组成。在VCaP 异种移植斑马鱼中,8.3 μmol·L-1的化合物A031 表现出55%的肿瘤生长抑制率,与恩杂鲁胺相当,药代动力学性质良好,毒性较低[58]。
4.2.2 基于羟脑苷脂配体的蛋白降解靶向嵌合体Takwale 等[59]通过使用芳氧基环丁胺AR 拮抗剂和羟脑苷脂(cereblon,CRBN)配体得到TD-802(45),该化合物是首个报道的具有CRBN 配体的AR PROTAC。TD-802 在LNCaP 细胞中的DC50为12.5 nmol·L-1,在体内异种移植物小鼠模型中能有效抑制肿瘤生长,并表现出良好的肝微粒体稳定性和体内药代动力学性质。
ARV-110(46)由阿维纳斯公司研发,经过对恩杂鲁胺和多种CRBN 配体及多种连接链的组合筛选,及进一步结构优化而得,为最早进入临床的AR PROTAC 分子[60]。在体内对恩杂鲁胺获得性和内在耐药模型中,经口给予3.0 mg·kg-1ARV-110,分别显示出70%和100%的肿瘤生长抑制率。ARV-110 在Ⅰ期临床试验中用于mCRPC 患者,具有良好的耐受性、安全性和药代动力学性质[61]。2022 年2 月,阿维纳斯公司披露ARV-110 在治疗mCRPC 的临床试验中,具有持续抗肿瘤活性和患者获益的证据,在携带ART878X/H875Y(T878X 为T878A 或T878S)突变肿瘤患者中,ARV-110 使46%患者的PSA 水平降低50%以上。目前,ARV-110 正在开展Ⅱ期临床试验[62]。
目前,另有多个AR PROTAC 已进入临床试验,其中阿维纳斯公司的ARV-766(47),相对于ARV-110,不仅对H875Y 和T878A 等多种突变体亚型具有降解能力,而且能更有效地降解与阿比特龙和其他AR 途径拮抗剂耐药相关的L702H 突变体亚型,并在动物模型中得以验证[63]。2021 年9月,ARV-766 在美国开展Ⅱ期临床试验,用于治疗mCRPC;其结构在2023 年4 月的美国癌症研究协会(American Association for Cancer Research,AACR)年会中被披露。此外,百时美施贵宝公司研发的CC-94676、冰洲石生物科技的AC-0176 以及海创药业研发的HP518 等PROTAC 分子都处于Ⅰ期临床试验阶段,用于治疗mCRPC[64]。
Kim 等[65]以比卡鲁胺为AR 配体,通过柔性链与CRBN 配体连接,设计并合成了一系列AR PROTAC。其中化合物48 能以剂量和时间依赖性的方式降解AR 蛋白(LNCaP:DC50=5.21 µmol·L-1)。另一系列的AR PROTAC 以恩杂鲁胺衍生物为AR配体,通过不同的三氮唑片段连接CRBN 配体,其中化合物49 具有优良的AR 结合亲和力(85%)和AR 降解活性[66]。
Han 等[67]以芳氧基环丁胺为AR 配体,采用含哌嗪的连接链与CRBN 配体沙利度胺相连,分别得到ARD-2128(50)和ARD-2585(51)。ARD-2128 在VCaP 和LNCaP 细胞中的DC50分别为0.28和8.3 nmol·L-1,能抑制AR 调控基因,经口给药可有效降低肿瘤组织中AR 蛋白,有效抑制小鼠肿瘤生长,且毒性较低[68]。ARD-2585 的降解活性更高,在VCap 和LNCaP 细胞中的DC50均低于0.10 nmol·L-1。ARD-2585 比恩杂鲁胺可更有效地降解全长AR 和ARVs,经口生物利用度(小鼠)达51%,体内疗效优于恩杂鲁胺,颇具临床应用潜力。
AR PROTAC 为CRPC 的治疗提供了新的治疗策略,但AR PROTAC 的相对分子质量较大,成药性亟需提升,实现良好的口服生物利用度具有挑战性。此外,AR PROTAC 多数作用于LBP 口袋,对于缺乏LBD 的ARVs 无效。因此,后续的AR PROTAC 的研究方向将聚焦解决上述问题。
5 糖皮质激素受体及其拮抗剂的应用
5.1 糖皮质激素受体的结构与功能
人源GR基因位于5 号染色体上,由9 个外显子组成。如图7 所示,GR 主要包含4 个区域,NTD 域由外显子2 编码,该区域主要负责转录激活功能以及和辅助调节因子结合;DBD 域由外显子3和4 编码,其结构上包含了2 个锌指结构识别DNA上的糖皮质激素反应元件(glucocorticoid-responsive elements,GREs);外显子5~9 编码铰链区(H)和LBD 域,LBD 域包含1 个配体结合口袋(LBP),以及1 个AF2 结构域,以配体依赖的方式与辅助调节因子相互作用[69]。

图7 糖皮质激素受体的结构Figure 7 Structure of glucocorticoid receptor
5.2 糖皮质激素受体与去势抵抗性前列腺癌
在前列腺癌中,AR 与TLE3(transducin-like enhancer of split 3)会和GR 的增强子相结合;另外,多梳抑制复合物2(polycomb repressive complex 2,PRC2)会导致组蛋白3 上的第27 位赖氨酸的三甲基化(trimethylation of lysine 27 on histone 3,H3K27me3),并通过zeste 基因增强子同源物2(enhancer of zeste homolog 2,EZH2)沉积到GR 启动子和增强子上,两者共同抑制GR 的表达[70]。在早期前列腺癌中,GR 表现为下调趋势,且在AR 激活的情况下,GR 可能发挥抑制肿瘤发生发展的作用。而在CRPC 患者中,恩杂鲁胺等AR 拮抗剂的使用,使得AR 的表达受到抑制,并且在治疗过程中TLE3会出现表达缺失,进而上调GR 增强子上的H3K27乙酰化,使原先H3K27 沉积受到抑制,从而恢复GR的表达[70]。而GR 表达升高后,会与ARE 结合,共同调控一些经典的AR 靶基因,导致前列腺癌的生长和恩杂鲁胺耐药的发生。另外,有研究表明GR 介导的葡萄糖转运蛋白4(glucose transporter 4,GLUT4)上调与前列腺癌治疗中出现的恩杂鲁胺耐药性和交叉耐药性相关,抑制GR 或GLUT4 后,可以减少葡萄糖的摄取,改善癌细胞的耐药性[71]。目前研究认为,GR 和AR 既存在协同作用,也存在对抗作用,其作用与肿瘤进展关系密切[72]。综上,GR 具有成为克服CRPC 耐药治疗关键靶点的潜力,具有器官靶向性的GR 拮抗剂及GR/AR 双重拮抗剂(结构式见图8)可能在治疗中更具有优势。
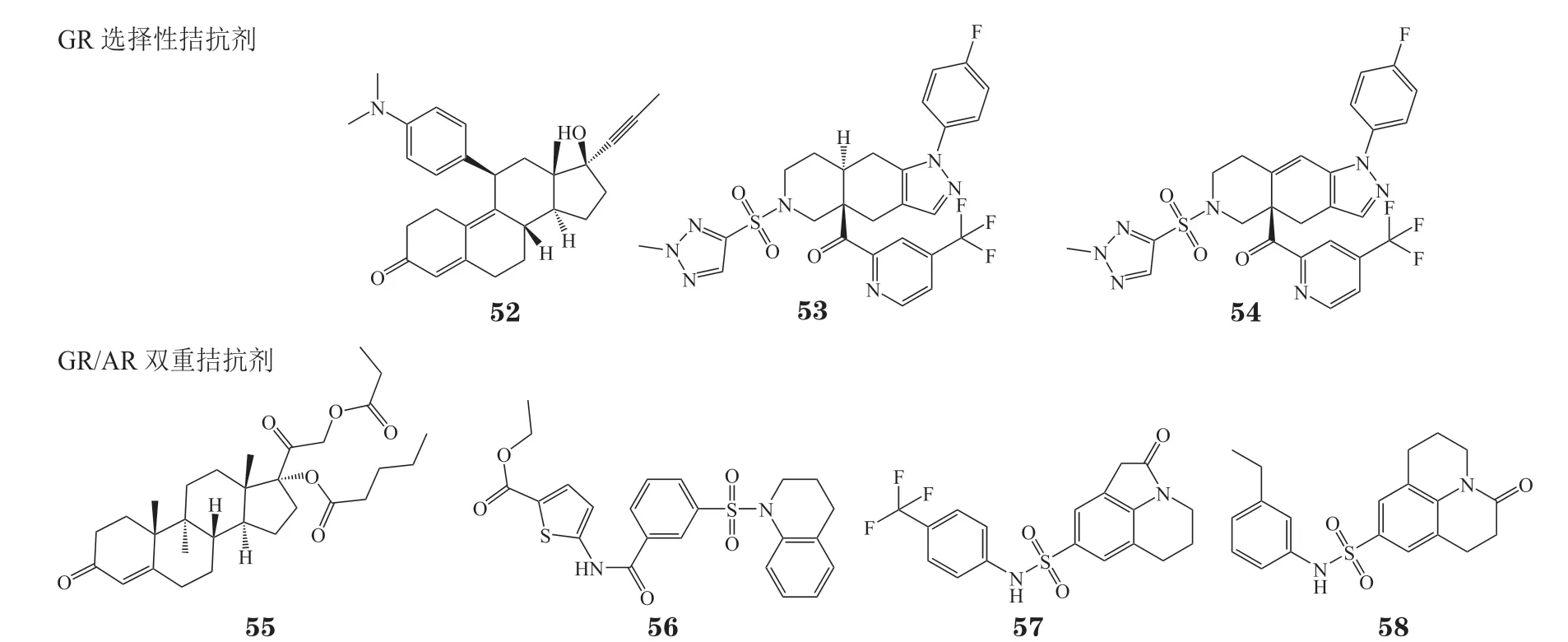
图8 糖皮质激素受体选择性拮抗剂和糖皮质激素受体/雄激素受体双重拮抗剂的化学结构式Figure 8 Chemical structures of glucocorticoid receptor antagonists and glucocorticoid receptor/androgen receptor dual antagonists
5.3 针对糖皮质激素受体过表达的治疗
5.3.1 联合糖皮质激素受体拮抗剂用药治疗去势抵抗性前列腺癌研究发现GR 拮抗剂可恢复耐药细胞对恩杂鲁胺的敏感性,从而提出联合用药治疗因GR 过表达引起的CRPC 耐药。2013 年,米非司酮(mifepristone,52)和恩杂鲁胺联用治疗CRPC 的临床试验(临床试验编号:NCT 02012296)启动,但因疗效不明显而终止[72]。2018 年,已开展GR 选择性拮抗剂exicorilant(53)和relacorilant(54)分别与恩杂鲁胺联用治疗CRPC 的临床试验(临床试验编号:NCT03437941,NCT03674814),但目前临床数据尚未公布。
5.3.2 雄激素受体/糖皮质激素受体双重拮抗剂联用方式治疗因GR 过表达而引起的CRPC 耐药具有一定的意义,但药物-药物相互作用的存在使临床应用受限。因此,靶向AR/GR 的双重拮抗剂是治疗GR过表达CRPC 的新方向。
对脱氧可的松结构改造所得的化合物CB-03-10(55)能有效抑制AR 和GR 的转录活性,对LNCaP 细胞和恩杂鲁胺耐药LNCaP-EnzaR 细胞均有效,并能抑制AR/GR 下游靶基因和蛋白的表达。在小鼠异种移植LNCaP 细胞模型中,CB-03-10 的活性与紫杉醇相当,具有良好的开发潜力[73]。
基于AR LBP 蛋白虚拟筛选发现的化合物Z19(56)对AR 和GR 均具有良好的转录抑制活性,IC50分别为2.03 和2.50 μmol·L-1。Z19 能抑制表达AR 突变22Rv1 细胞系和AR 阴性PC-3 细胞系生长,降低GR 和AR 信号下游蛋白和mRNA 的水平,有效抑制CRPC 耐药肿瘤的增殖。生物膜干涉实验和分子对接研究表明,该化合物可能靶向AR 和GR的LBP 口袋,与内源性配体竞争性结合AR/GR 而发挥拮抗作用[74]。
基于AR LBP 蛋白虚拟筛选发现的化合物H18(57)具有一定的AR 拮抗活性,经分子动力学模拟研究及结构优化得到AR/GR 双重拮抗剂HD57(58),该化合物对AR 和GR 转录抑制IC50分别为0.39 和17.81 μmol·L-1。此外,该分子对大部分AR 突变体的抑制活性与达洛鲁胺相当,并能抑制AR 下游靶基因PSA的表达和AR 核易位[75]。
6 结语与展望
AR 和GR 与前列腺癌的发生、发展密切相关,是抗前列腺癌新药研发的重要靶标。不过,目前已上市的AR 拮抗剂均靶向AR 的LBP 位点,且存在交叉耐药性,限制了临床上的使用。因此,靶向AR 的非LBP 位点引起越来越多的关注,包括AR LBD 上的AF2 和BF3 位点,AR 的DIP 口袋,AR的NTD 及DBD 域等,尤其是后2 个靶点,有望解决ARVs 缺乏LBD 域的问题。另一方面,近年来AR PROTAC 已成为AR 靶向治疗的热点,ARV-110和ARV-766 已分别进入Ⅱ期临床试验,且疗效明显,前景光明。另外,基于AR DBD 抑制剂的PROTAC(如MTX-23),为治疗AR-V7 导致的CRPC 亚群提供了新的解决方案。GR 拮抗剂以及AR/GR 双重拮抗剂具有独特的作用机制,在CRPC 治疗领域预期有更广阔的前景。
综上所述,靶向核受体AR 和GR 是临床治疗前列腺癌的重要策略,虽然耐药突变、PROTAC 生物利用度低等问题亟需解决,但相信随着计算机辅助药物设计(computer aided drug design,CADD)、人工智能(artificial intelligence,AI)和PROTAC技术的进一步发展,将会有更多的靶向核受体抗前列腺癌新药进入临床,为广大患者提供更好的治疗选择。
