抗前列腺癌相关核受体及小分子药物研究进展
2023-10-20卢志芳吴桐吴锡山李俊骅张岩刘洋翟鑫许永
卢志芳 ,吴桐 ,吴锡山,李俊骅,张岩,刘洋,翟鑫*,许永**
(1.沈阳药科大学 基于靶点的药物设计与研究教育部重点实验室,辽宁 沈阳 110016;2.中国科学院广州生物医药与健康研究院化学生物学与药物研究中心,广东 广州 510530)
前列腺癌(prostate cancer,PCa)指发生于前列腺的上皮恶性肿瘤,其发病率在男性癌症中排名第2,严重影响男性的健康。GLOBOCAN 2020 发布的调查统计表明,新增的前列腺癌患者多达140万人,其中约37.5 万人死亡[1],预计新增和死亡人数将在2040 年达到新的高峰。前列腺癌的致病因素多样且复杂,年龄、家族遗传史、不良生活方式、体内激素水平、代谢调节和肠道微生物群等都可能对前列腺癌的发生发展产生影响[2-4]。
雄激素及雄激素受体(androgen receptor,AR)信号通路在前列腺癌的发生发展中发挥着重要作用,目前大部分治疗前列腺癌的手段都围绕AR信号通路展开[5]。但由于AR基因的突变和扩增、雄激素生物合成和AR 辅助因子的改变以及剪接变体的表达,癌细胞几乎不可避免地产生耐药,最终发展为去势抵抗性前列腺癌(castration-resistant prostate cancer,CRPC)[6]。此类癌症患者预后极差,治疗颇为棘手。虽然患者受益于第2 代抗雄激素药物AR 拮抗剂恩杂鲁胺(enzalutamide)和细胞色素P450 家族17 亚家族A 成员1(cytochrome P450 family 17 subfamily A member 1,CYP17A1)抑制剂阿比特龙(abiraterone),但均会进一步产生二次耐药[7]。因此,亟需发现治疗CRPC 的新机制、新靶点,为临床有效克服CRPC 提供新的治疗策略。
CRPC 的发生机制主要包括AR 依赖性机制和AR 非依赖性机制[8-10]。AR 依赖性机制包括AR基因扩增与过表达、AR基因突变、AR 共调节因子表达的改变等。在AR 依赖性机制中AR 敏感性增高,进一步导致雄激素信号通路的持续激活。AR 非依赖性机制包括磷脂酰肌醇3 激酶-蛋白激酶B-哺乳动物雷帕霉素靶蛋白(phosphoinositide 3 kinase-protein kinase B-mammalian target of rapamycin,PI3K-AKTmTOR)信号通路、Wnt/β 联蛋白(β-catenin)信号通路、旁路途径和干细胞途径等,这些机制同样与AR 信号通路的激活紧密相关。综上所述,前列腺癌转变为晚期CRPC 的过程中,有诸多细胞内信号通路参与其中,包括AR 信号通路和非AR 信号通路,其中AR信号通路是CRPC发展过程中的主要通路。
考虑到核受体AR 在前列腺癌中的关键地位,且与其他核受体之间存在相互作用,药物学家们针对其他核受体也进行了研究,以期发现新的抗前列腺癌的核受体药物靶标。
核受体由氨基末端(A/B)域、DNA 结合域(DNA-binding domain,DBD)、铰链区和配体结合域(ligand-binding domain,LBD)4 个主要功能域构成。其中LBD 区域可以结合小分子配体,还能提供与共调节因子相互作用的位点,使核受体产生多样化的基因调控作用。核受体与生物的生长、发育、代谢等生理过程密切相关[11-12]。
目前已有研究表明,除AR 外还有众多核受体也与前列腺癌相关。这些核受体包括肝X 受体(liver X receptor,LXR)α/β、维甲酸受体相关孤儿受体γ(retinoic acid receptor-related orphan receptor γ,RORγ)、维甲酸受体γ(retinoic acid receptor γ,RARγ)、过氧化物酶体增殖物激活受体γ(peroxisome proliferator-activated receptor γ,PPARγ)、法尼醇X受体(farnesoid X receptor,FXR)、鸡卵清蛋白上游启动子转录因子Ⅱ(chicken ovalbumin upstream promoter transcription factor II,COUP-TFII)、核受体无尾蛋白(tailless like protein,TLX)、睾丸受体4(testicular receptor 4,TR4)、雌激素受体(estrogen receptor,ER)α/β、雌激素 相关受体(estrogenrelated receptor,ERR)α/γ、肝脏受体同源物-1(liver receptor homolog-1,LRH-1)、类固醇生成因子-1(steroidogenic factor-1,SF-1)、生殖细胞核因子(germ cell nuclear factor,GCNF)、小异二聚体伴侣(small heterodimer partner,SHP)、X 染色体基因1上剂量敏感的性别反转-先天性肾上腺发育不良关键区域(dosage-sensitive sex reversal-adrenal hypoplasia congenita-critical region on the X chromosome gene 1,DAX1)、糖皮质激素受体(glucocorticoid receptor,GR)、孕烷X 受体(pregnane X receptor,PXR)、维生素D 受体(vitamin D receptor,VDR)和盐皮质激素受体(mineralocorticoid receptor,MR)等。本文将对这些核受体在前列腺癌中的研究进展进行综述。相关核受体在前列腺癌中的作用机制和靶向核受体的抗前列腺癌小分子化合物分别见图1 和表1[13-30]。
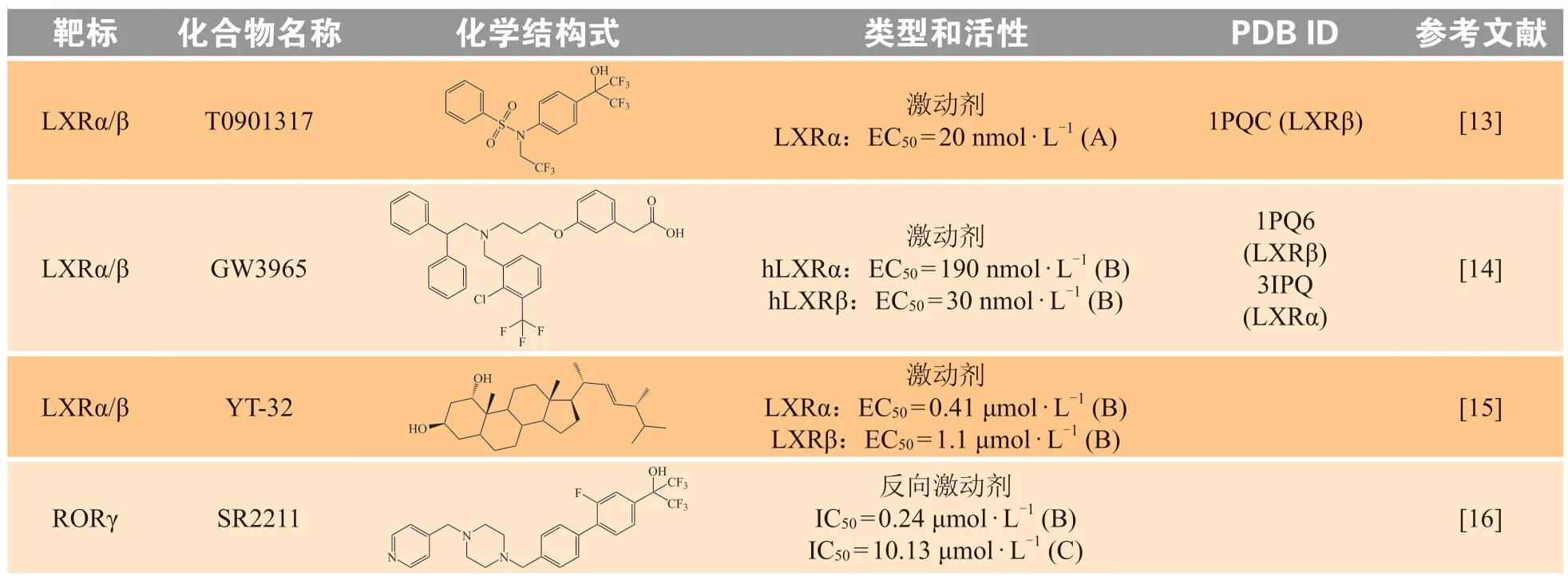
表1 靶向核受体的抗前列腺癌小分子化合物Table 1 Small molecule compounds targeting nuclear receptos for the treatment of prostate cancer
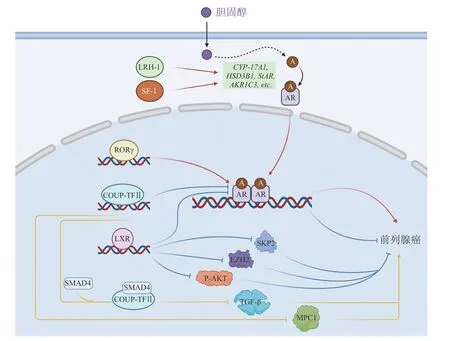
图1 核受体LRH-1,SF-1,RORγ,COUP-TFII 和LXR 在前列腺癌中的作用机制Figure 1 The action mechanisms of nuclear receptors LRH-1,SF-1,RORγ,COUP-TFII and LXR in prostate cancer
1 肝X 受体α/β
1.1 肝X 受体α/β 与前列腺癌
LXR 属于核受体第Ⅰ家族,为配体依赖性转录因子,包括LXRα 和LXRβ 2 种亚型。LXRα 在肝脏中高度表达,同时在肠、脂肪、肾和肾上腺等参与胆固醇代谢的器官和组织中也有表达,而LXRβ 在体内各组织中都有表达[31]。
Viennois 等[32]发现LXRα 可以调控前列腺中的雄激素及其受体信号通路。还有研究表明LXRα 激动剂T0901317 可以抑制雄激素依赖性基因的转录激活和前列腺特异性抗原(prostate specific antigen,PSA)的表达,同时抑制雄激素与AR 的结合[33]。此外,β 联蛋白也参与AR 的功能调节,可增强雄激素刺激的AR 转录活性。而LXRα 的激活可以通过抑制β 联蛋白来抑制细胞增殖和AR 转录活性[34]。因此,LXRα 可以通过调控AR 的活性在前列腺癌的发生发展中发挥作用(见图1)。
另一方面,LXR的激活还可以抑制雄激素的活性。Lee 等[35]发现LXR 通过诱导磺基转移酶家族2A 成员1(sulfotransferase family 2A member 1,SULT2A1)的表达来降低雄激素的活性,同时LXR 的激活也抑制了前列腺中类固醇硫酸酯酶(steroid sulfatase,STS)的表达,阻止磺化雄激素局部转化为活性代谢物,进而抑制雄激素依赖性前列腺癌细胞的增殖。
Fukuchi 等[36]发现T0901317 可以抑制前列腺癌细胞的增殖,且造成细胞中细胞周期蛋白依赖性激酶抑制因子p27Kip-1(cyclin-dependent kinase inhibitor p27Kip-1,p27)表达增加、S 期激酶相关蛋白2(S-phase kinase-associated protein 2,SKP2)表达减少,这一结果可能是通过影响细胞中LXR 的信号通路造成的(见图1)。研究显示,经口给予T0901317 可抑制裸鼠LNCaP 肿瘤的生长。
Pommier 等[37]发 现T0901317 还可以通过下调AKT 的磷酸化来使内质膜片层的脂筏变小变薄,从而诱导LNCaP 细胞凋亡,这实际上是LXR诱导ATP 结合盒转运体G1(ATP binding cassette transporter G1,ABCG1)的上调而后增加胆固醇的反向转运使得肿瘤细胞的胆固醇含量下降所导致。他们还发现在LXRα/β-/-双基因敲除小鼠中,zeste 基因增强子同源物2(enhancer of zeste homolog 2,EZH2)的表达增加导致了肿瘤抑制基因微精蛋白β(microseminoprotein beta,MSMB)和同源框蛋白Nkx-3.1(homeobox protein Nkx-3.1,Nkx3.1)的表达下调,从而使得体内的胆固醇酯异常堆积和前列腺上皮内瘤变(见图1)[38]。
1.2 靶向肝X 受体α/β 的抗前列腺癌小分子化合物
LXR 小分子激动剂大致分为2 类,一类是天然的激动剂,包括氧化甾醇、22(R)-羟基胆固醇、植物甾醇等;另一类是合成的激动剂,如T0901317,GW3965,YT-32,GW6340 等(见表1)。其中Tularik 公司研发的化合物T0901317[13]和葛兰素史克(GSK)公司研发的化合物GW3965[14]是较早的人工合成的LXR 激动剂,二者与天然化合物相比具有更高的亲和力。化合物T0901317 可以与LXRα 较好结合,其半数效应浓度(median effective concentration,EC50)为20 nmol·L-1,化合物GW3965 可以有效激活LXRα(EC50=190 nmol·L-1)和LXRβ(EC50=30 nmol·L-1)。科研人员将这2 个化合物作为分子探针进一步阐明了LXR 在前列腺癌中的作用机制。在后续研究中发现T0901317 和GW3965 等LXR 激动剂会活化肝脏的LXR,导致肝脏的甘油三酯升高,这限制了二者在临床上的应用。为避免这一副作用,科研人员开发了一些具有选择性的LXR 激动剂,如GW6340 和YT-32 等,其中YT-32 是与麦角甾醇和油菜甾醇相关的植物甾醇衍生物之一,对LXRα 的EC50为0.41 μmol·L-1,YT-32 可以在不增加血浆甘油三酯水平的前提下抑制肠道对于胆固醇的吸收[15]。
2 维甲酸受体相关孤儿受体γ
2.1 维甲酸受体相关孤儿受体γ 与前列腺癌
RORγ 是核受体第Ⅰ家族中一类重要的孤儿受体。它可调控相关基因的表达,在生长发育、免疫和代谢等生命过程中发挥关键作用。RORγ 还与自身免疫性疾病和癌症等多种疾病密切相关,是重要的药物靶标[39]。
笔者课题组研究发现,核受体RORγ 作为关键驱动因子,在AR 表达、转录、激活等过程中发挥重要作用。由于抑制RORγ 则可抑制全长的AR(见图1)、LBD 突变的AR 以及缺少LBD 的雄激素受体剪接变体7(androgen receptor splice variant 7,AR-V7)等多种形式AR 的表达,故而RORγ 抑制剂可用于CRPC 的治疗。因此,RORγ 可作为新的抗前列腺癌药物靶标,开发其抑制剂将成为治疗前列腺癌的新策略[40]。
2.2 靶向维甲酸受体相关孤儿受体γ 的抗前列腺癌小分子化合物
目前,已有多个RORγ 小分子抑制剂被用于前列腺癌的研究(见表1)。笔者团队于2016 年发现RORγ 小分子抑制剂SR2211[16]和XY018[17]可以在体内外有效抑制前列腺癌细胞的增殖[40]。在化合物XY018 的结构基础上继续优化,获得了2 个高活性和高选择性的化合物XY101[17]和XY123[16]。化合物XY101 和XY123 均可有效抑制RORγ 的转录活性,半数最大抑制浓度(half maximal inhibitory concentration,IC50)分别为0.03 和0.064 μmol·L-1。它们还具有良好的代谢稳定性和药代动力学性质,口服生物利用度分别为59%和32%。在体外,这2个化合物可以抑制多种前列腺癌细胞的增殖与克隆形成,同时可有效抑制前列腺癌细胞中AR,AR-V7和AR 调控的下游基因的表达。在体内,它们均可有效抑制小鼠体内前列腺肿瘤生长。
Zou 等[18]发现天然产物熊果酸在前列腺癌细胞中可有效抑制RORγ 的转录活性。熊果酸可有效抑制C4-2B 和22Rv1 细胞的增殖,IC50分别为8.51 和7.76 μmol·L-1。熊果酸目前处于Ⅱ期临床研究,但其临床试验靶点并不针对RORγ,临床适应证为肝癌、胃癌和结直肠癌等。Zheng 等[19]发现天然产物洋橄榄叶素也可以作为RORγ 的小分子抑制剂。在表面等离子共振(surface plasmon resonance,SPR)实验中,洋橄榄叶素可与RORγ-LBD 较强地结合,其解离常数(dissociation constant,Kd)为5.05×10-6mol·L-1。洋橄榄叶素可以在纳摩尔浓度抑制前列腺癌细胞22Rv1,VCaP 和LNCaP 的增殖。在22Rv1异种移植的小鼠模型中,洋橄榄叶素也可显著抑制肿瘤的生长。
3 维甲酸受体γ
3.1 维甲酸受体γ 与前列腺癌
RARγ 位于细胞核内,属于核受体第Ⅰ家族成员。它可在体内与全反式维甲酸(all trans retinoic acid,ATRA)结合从而激活维甲酸信号转导途径,调控下游靶基因的转录,维持机体的生理稳态[41]。
ATRA 可以通过调控RARγ 影响早期前列腺前体细胞的发育。前列腺癌组织中含有非常低水平的ATRA(在10-9mol·L-1范围内),这至少比邻近的正常前列腺细胞低1 个数量级。低浓度的ATRA 可通过激活RARγ 来抑制前列腺癌细胞分化成脂肪细胞,促进前列腺癌细胞的增殖与存活。抑制RARγ可以作为前列腺癌治疗的新策略[21]。
3.2 靶向维甲酸受体γ 的抗前列腺癌小分子化合物
目前,用于前列腺癌治疗的RARγ 抑制剂较少。Hammond 等[20]发现,RARγ 抑制剂AGN194431 可以抑制前列腺癌细胞的增殖,但该化合物对RARγ的结合(Kd=70 nmol·L-1)没有选择性,对RAR 家族的另2 个亚型RARα 和RARβ 也有结合活性。经后续化学结构修饰获得了RARγ 的选择性小分子抑制剂AGN205728[21]。该化合物可较好地与RARγ 结合,ED50为3 nmol·L-1。AGN205728 可以阻断雄激素依赖性和非雄激素依赖性前列腺癌细胞的增殖,并诱导半胱天冬酶非依赖性细胞凋亡。单独使用AGN205728 或与细胞毒性化疗联合使用可用于治疗晚期前列腺癌(见表1)。
4 过氧化物酶体增殖物激活受体γ
4.1 过氧化物酶体增殖物激活受体γ 与前列腺癌
PPARγ 属于核受体第Ⅰ家族,是脂肪生成的主要调节因子,其天然配体大多为各类脂肪酸[42-43]。
研究显示,PPARγ 与前列腺癌具有关联性,其促癌作用主要通过3 种机制通路实现:1)通过脂肪酸合成酶(fatty acid synthase,FASN)和ATP 柠檬酸裂解酶(ATP citrate lyase,ACLY)介导脂肪酸合成,促进前列腺癌细胞的生存;2)通过丝氨酸/苏氨酸蛋白激酶3(AKT serine/threonine kinase 3,AKT3)、过氧化物酶体增殖物激活受体γ 共激活因子1α(PPARγ coactivator 1 alpha,PGC1α)和染色体维持蛋白1(chromosome maintenance region 1,CRM1)组成的信号轴驱动线粒体的生物合成,促进肿瘤的生长和转移;3)在CRPC 中,配体诱导的PPARγ 激活介导了AR 信号的激活[44]。
4.2 靶向过氧化物酶体增殖物激活受体γ 的抗前列腺癌小分子化合物
目前,已有一些靶向PPARγ 的抗前列腺癌小分子化合物被报道(见表1)。例如,T0070907 能够下调FASN,并通过PPARγ-AR 途径抑制前列腺肿瘤的生长[22,45]。Almahmoud 等[46]通过虚拟筛选得到了一系列抗前列腺癌的PPARγ 抑制剂。其中,基于变构位点筛选出的小分子抑制剂相较于正构位点具有更好的抑制活性,在细胞增殖抑制(LNCaP 细胞)和荧光素酶报告基因实验中表现出微摩尔抑制活性。另外,吡格列酮和罗格列酮等噻唑烷二酮类的PPARγ 激动剂可以不依赖PPARγ 对前列腺癌产生治疗作用。它们通过对C-X-C 基序趋化因子受体4(C-X-C motif chemokine receptor 4,CXCR4)/C-X-C基序趋化因子配体12(C-X-C motif chemokine 12,CXCL12)轴产生抑制作用,进而介导胞外信号调节激酶(extracellular signal-regulated kinase,ERK)的磷酸化来抑制前列腺癌的发展[47]。
5 法尼醇X 受体
FXR 是核受体第Ⅰ家族成员,能够被胆汁酸结合激活,进而调控多种代谢相关基因的表达。研究表明,FXR 对于前列腺癌表现出促癌与抑癌的双重作用。Kaeding 等[48]发现FXR 在LNCaP 细胞系中过表达,能够调节雄激素的代谢,是雄激素依赖性前列腺癌的重要诱因。与之相对的,Liu 等[49]观察到前列腺癌组织中的FXR 表达相比正常组织有所降低,而FXR 在前列腺组织的激活与过表达,能够上调第10 号染色体缺失的磷酸酶与张力蛋白同源基因(phosphatase and tensin homologue deleted on chromosome ten,PTEN)的表达并下调AKT 的磷酸化水平,从而产生抑癌作用。
目前,已报道的靶向FXR 的抗前列腺癌小分子化合物均为FXR 激动剂。其中,天然配体鹅去氧胆酸(chenodeoxycholic acid,CDCA)及人工合成的FXR 激动剂GW4064(见表1),均能抑制AKT 的磷酸化,从而抑制前列腺癌细胞的生长[23,49-50]。
6 鸡卵清蛋白上游启动子转录因子Ⅱ
6.1 鸡卵清蛋白上游启动子转录因子Ⅱ与前列腺癌
COUP-TFII 属于核受体第Ⅱ家族,是类固醇/甲状腺激素受体家族的重要转录因子。COUP-TFII在调节血管生成、神经元发育和代谢稳态等多种生物过程中发挥关键作用[51]。异常调控的COUP-TFII参与了包括癌症在内的许多病理过程。越来越多的研究表明,COUP-TFII 在前列腺癌、肺癌、结直肠癌和胰腺癌等多种类型的癌症中过表达[52],揭示了COUP-TFII 是潜在的抗癌靶点。
在PTEN缺失小鼠中,COUP-TFII 在前列腺上皮中的过表达加速了前列腺肿瘤的发展和癌细胞的转移。虽然PTEN缺失引起的转化生长因子-β(transforming growth factor-β,TGF-β)信号上调会形成生长屏障来抑制前列腺癌发展,但COUP-TFII可以结合并抑制母亲DPP 同源物4(mothers against decapentaplegic homolog 4,SMAD4)的转录活性,抵消TGF-β 信号诱导形成的生长屏障以促进前列腺癌的发展(见图1)[53]。
线粒体丙酮酸载体1(mitochondrial pyruvate carrier 1,MPC1)和MPC2 可以在细胞内形成转运复合体来控制丙酮酸向线粒体的转运。COUP-TFII通过抑制前列腺癌细胞中MPC1 的表达,扰乱转运蛋白的功能,导致代谢转变,增加了糖酵解,进而促进癌细胞的生长和转移(见图1)[54]。
Song 等[55]发现,COUP-TFII 过表达可抑制雄激素依赖性前列腺癌细胞的增殖。COUP-TFII是AR 的共抑制因子,能够同时与AR 的DBD 和LBD 结合,并破坏AR 的N/C 端相互作用。此外,COUP-TFII 还可以与AR 共激活因子竞争,以调节AR 的转录激活,并抑制AR 向含有雄激素响应元件(androgen response element,ARE)的靶启动子的募集(见图1)。
6.2 靶向鸡卵清蛋白上游启动子转录因子Ⅱ的抗前列腺癌小分子化合物
COUP-TFII 是治疗前列腺癌的重要靶点,其活性受到配体的调控。目前已针对COUP-TFII 开发了一些小分子抑制剂,如CIA1 和CIA2(见表1)。CIA1 和CIA2 可直接与COUP-TFII 的LBD 结合,破坏COUP-TFII 与转录调控因子的相互作用,从而抑制COUP-TFII 对靶基因的调控活性以达到抗前列腺癌的作用。CIA1 和CIA2 能够抑制多种前列腺癌细胞系的生长,IC50分别为1.2~ 7.6 和2.2~ 10.2 μmol·L-1,而对正常前列腺细胞的生长几乎没有影响。在药代动力学实验中,CIA1 在血浆中的浓度1小时内下降到原来的1/10,而在脂肪、肝脏和睾丸等组织中含量较高,这表明CIA1 在体内的清除率及组织选择性有待提高[24]。
7 核受体无尾蛋白
TLX 是核受体第Ⅱ亚家族的成员,最早作为果蝇无尾蛋白的同源类似物被发现。TLX 是进化保守的核受体,通常活跃于增殖的神经祖细胞,调节成体神经干细胞的自我更新,在胚胎和成人神经发生中起着关键作用[56]。
已有研究表明,在多种CRPC 细胞系中,TLX的表达水平显著上调。TLX 的敲除能够增强雄激素剥夺和抗雄激素治疗对前列腺癌细胞的增殖抑制能力。同时,TLX 还可以通过直接与AR 启动子结合,招募赖氨酸脱甲基酶1(lysine-specific demethylase 1,LSD1)、组氨酸去乙酰化酶1(histone deacetylase 1,HDAC1)和HDAC3 来抑制AR 的转录激活[57]。此外,Wu 等[58]研究发现,TLX 通过对衰老调控基因细胞周期蛋白依赖性激酶抑制因子1A(cyclin-dependent kinase inhibitor 1A,CDKN1A,p21)和沉默调节蛋白1(sirtuin 1,SIRT1)的差异性共调控(differential co-regulate),抑制癌基因诱导的衰老,从而促进前列腺癌的发生发展。因此,TLX 可作为前列腺癌的潜在治疗靶点。
目前,已有一些TLX 小分子调节剂被开发,但它们的适应证主要集中于神经调节方面,针对抗前列腺癌的活性,还有待进一步的评价[56]。
8 睾丸受体4
TR4 是核受体第Ⅱ家族的成员,于1994 年在人类前列腺和睾丸cDNA 库中被克隆出来,其内源性配体目前仍未知。TR4 可能作为脂质传感器在能量稳态、神经元发育和生育中发挥重要作用。近年来,研究发现,TR4 在癌症的不同阶段发挥不同的作用。在前列腺癌早期阶段,TR4 扮演了肿瘤抑制因子的角色,它通过促进DNA 修复和维持基因组的完整性来抑制前列腺癌的发生发展[59]。但在PPARγ 缺失的情况下,TR4 会转变成致癌因子,它通过增加肿瘤干细胞数量和增强上皮-间充质转化促进前列腺癌的发生[60]。另外,TR4 也作为促癌因子促进前列腺癌的迁移和侵袭[61]。TR4 还会导致前列腺癌对化疗和放疗的抵抗。研究发现,TR4 高表达的前列腺癌细胞对依托泊苷化疗有抵抗作用[62]。同时,TR4 的上调也可以增强前列腺癌细胞的耐辐射能力。前期研究发现,视黄醇可以作为TR4 配体激活其功能,但目前TR4小分子调节剂的抗前列腺癌活性仍需进一步研究[63]。
9 雌激素受体α/β
ER 为配体依赖的转录因子,属于核受体第Ⅲ家族。它与雌激素响应元件结合,调节靶器官的生长、分化和发育。ER 具有ERα 和ERβ 2 种亚型,它们在结构上高度相似,但在细胞和组织中的表达不同[64]。在前列腺组织中,ERα 主要表达于前列腺间质细胞,而ERβ 主要表达于前列腺上皮细胞和间质细胞,介导前列腺上皮细胞的分化和细胞形态的维持。在前列腺癌中,ERα 作为致癌因子高表达,促进癌细胞的增殖、侵袭和迁移,并随着肿瘤的进展而逐渐增加其表达水平[65]。ERβ 也是重要的肿瘤调节因子,通过直接或间接的方式调节细胞的增殖和代谢。ERβ 可以与AR 相互作用,在转录或转录后水平上调节AR 信号,间接与特异性蛋白-1(specificity protein-1,SP-1)、核因子-κB(nuclear factor-κB,NF-κB)相互作用,从而影响前列腺癌的发展。此外,多种调节因子也可以通过调节ERβ 的状态来控制前列腺癌的进展[66]。目前已有一些靶向ER 的小分子化合物进入临床并用于前列腺癌的治疗,如选择性ERα 抑制剂托瑞米芬[25]和混合雌激素激动剂/抑制剂雷洛昔芬[26]等。其中,托瑞米芬处于Ⅲ期临床研究,雷洛昔芬处于Ⅱ期临床研究(见表1)。
10 雌激素相关受体α/γ
ERR 属于核受体第Ⅲ家族,包含α,β,γ 3 种亚型,其DBD 和LBD 与ER 高度同源,但不与雌激素结合,属于孤儿核受体[67]。
ERRα 在前列腺癌的发展中表现出2 个方面的致癌作用:其一,ERRα 的过表达上调了缺氧诱导因子-1α(hypoxia inducible factor-1α,HIF-1α)的表达,并且ERRα 的激活功能区2(activation function 2,AF2)与HIF-1α 相互作用又能避免HIF-1α 被泛素化,在上述2 种途径共同作用下,HIF-1α 的表达被维持在较高的水平,从而使得前列腺癌细胞获得对缺氧环境更高的耐受性;其二,ERRα 能够直接激活细胞色素P450家族11亚家族A成员1(cytochrome P450 family 11 subfamily A member 1,CYP11A1)和醛酮还原酶家族1 成员C3(aldo-keto reductase family 1 member C3,AKR1C3)2 种关键的雄激素合成酶,促进二氢睾酮的生成,从而激活AR 信号。ERRα 抑制剂XCT790 能够同时抑制上述2 种作用机制,从而产生抗前列腺癌作用[68-69]。
与ERRα 不同,ERRγ 在前列腺癌中表现抑癌作用。Yu 等[70]研究表明,ERRγ 能够调控p21 和p27 的表达,将细胞周期阻滞在G1-S 期,抑制前列腺癌细胞的增殖。另一方面,Audet-Walsh 等[71]证明了激活AR 信号可以对ERRγ 产生抑制,提高线粒体活性,促进细胞增殖。目前尚未有针对前列腺癌的ERRγ 激动剂被报道。
2020 年,Schoepke 等[27]报道了可同时靶向ERRα/γ 的抑制剂SLU-PP-1072。SLU-PP-1072 具有不同于XCT790 的新颖母核,并能通过抑制Warburg 效应介导前列腺癌细胞的凋亡(见表1)。
11 肝脏受体同源物-1
11.1 肝脏受体同源物-1 与前列腺癌
LRH-1 是核受体第Ⅴ家族成员,参与细胞的增殖发育、代谢活动等重要生理过程。目前,已有研究证明,LRH-1 是糖尿病、心血管疾病以及多种癌症的潜在治疗靶点[72-73]。
LRH-1 作为孤儿受体,能够在不结合配体的情况下保持激活构象。而近年来的研究表明,多种磷脂可能是其内源性配体。同时,不同配体的结合会导致LRH-1 LBD 的H12 的重新定位,驱动共调节因子的招募[74]。
Xiao 等[75]研究发现,LRH-1 在CRPC 中高表达,通过激活若干关键的类固醇生成酶,可以促进雄激素的从头生物合成,进而提升癌细胞内雄激素水平和AR 信号强度,从而加深前列腺癌的去势抗性(见图1),而使用LRH-1 抑制剂能够减弱前列腺癌的去势抵抗。LRH-1 可作为前列腺癌治疗的潜在靶点。
11.2 靶向肝脏受体同源物-1 的抗前列腺癌小分子化合物
目前,针对前列腺癌的LRH-1 反向激动剂的研究还十分有限,尚未有LRH-1 与反向激动剂结合的共晶结构被报道。Busby 等[28]在2011 年通过筛选发现了LRH-1 的反向激动剂ML179 和ML180(见表1)。报告基因实验证实,ML179 和ML180 对LRH-1 介导的细胞色素P450 家族成员19(cytochrome P450 family 19,CYP19)芳香化酶表达表现出抑制作用(ML179:IC50=320 nmol·L-1,ML180:IC50=3 700 nmol·L-1)。后续研究进一步证实,ML180 可以剂量依赖性地抑制VCaP 细胞中关键类固醇生成酶基因的表达,并抑制VCaP 和LNCaP 细胞的增殖[75]。然而,ML180 似乎并不与LRH-1 的LBD 结合,而是与LRH-1 全长蛋白发生作用。这提示ML180 可能结合于LRH-1 的全新结合位点[76]。
12 类固醇生成因子-1
SF-1 属于核受体第Ⅴ家族,与LRH-1 高度同源,是主要存在于类固醇生成组织的转录因子。SF-1 可以驱动与胆固醇代谢和类固醇激素转化相关基因的表达,还可以促进细胞的增殖和存活,但相关机制还不明确[77]。
Lewis 等[78]首先阐明了SF-1 对前列腺癌的促进作用。SF-1 在正常前列腺细胞中不表达,但在侵袭性前列腺癌细胞系中表达。其存在能够诱导关键的类固醇生成酶基因的表达,促进雄激素的生物合成(见图1)。同时,SF-1 还能够通过维持细胞中心体稳态,对前列腺癌细胞的增殖产生促进作用。经由SF-1 介导,异种移植前列腺癌细胞能够在类固醇缺乏的环境中生长,证明了SF-1 与CRPC 之间的相关性。目前,已有若干SF-1 小分子抑制剂(AC-45594,SID7969543,SID7970631)被报道(见表1),但这些小分子抑制剂的抗前列腺癌的活性仍有待研究[29]。
13 生殖细胞核因子
GCNF 又称维甲酸受体相关的睾丸相关受体,是核受体第Ⅵ家族中的孤儿受体。GCNF 是许多脊椎和无脊椎动物胚胎发育所必需的,可以抑制胚胎干细胞分化过程中的基因表达。近年来有研究表明,细胞中GCNF 的水平与前列腺癌的发展相关。GCNF在前列腺癌细胞中转录水平升高,沉默前列腺癌细胞中GCNF 的表达可使细胞周期阻滞在G0/G1期,从而显著降低前列腺癌细胞的侵袭和转移潜能。因此,GCNF 可能在前列腺癌的发展中发挥关键作用[79-80]。
14 小异二聚体伴侣
SHP 是缺乏DNA 结合域的核受体第0 家族成员,它可以与AR,ER,LRH-1 和DAX1 等核受体结合,发挥转录共抑制因子的作用,是重要的转录和代谢调节因子[81]。
有研究发现SHP 在多种人前列腺癌细胞系中表达下调,SHP 的下调与微RNA-141(microRNA-141,miR-141)的上调有关。因此,可以通过抑制miR-141 功能去增强SHP 的表达来减弱人前列腺癌细胞中AR 的转录活性[82]。SHP 还可能通过与AR 共激活肽竞争来抑制AR 的转录活性[83]。
异硫氰酸苯乙酯是许多可食用十字花科蔬菜的天然成分,可以通过下调前列腺癌细胞中的miR-141 和上调SHP 来抑制AR 的转录激活[82]。此外,人工合成的SHP 激动剂3-Cl-AHPC 也表现出对前列腺癌细胞DU-145 的增殖抑制活性,其IC50为0.5 μmol·L-1(见表1)[30]。
15 X 染色体基因1 上剂量敏感的性别反转-先天性肾上腺发育不良关键区域
DAX1 属于核受体第0 家族,在人类的性别决定和类固醇生成中发挥重要作用。DAX1 在下丘脑、垂体、肾上腺皮质以及性腺中的睾丸间质细胞、支持细胞(Sertoli cells)、卵泡膜和颗粒层细胞的发育过程中起关键作用。据报道,DAX1 还可抑制人类前列腺癌细胞系LNCaP 中的AR 活性,但对DAX1 在人类前列腺癌中的具体生物学作用仍不清楚[84-85]。目前尚无针对前列腺癌的DAX1 小分子化合物被报道。
16 与前列腺癌相关的其他核受体
GR 对前列腺癌的作用具有两面性。一方面,GR可以调控与前列腺癌细胞功能相关的转录因子来抑制肿瘤细胞的增殖、侵袭和迁移;另一方面,GR具有抗化疗和抗放疗的作用,可通过促进存活能力更强的静止癌细胞(quiescent cancer cell,QCC)的形成和上调氧化磷酸化水平等方式促进肿瘤细胞的存活[86]。此外,PXR[87],VDR[88]和MR[89]等核受体也被报道与前列腺癌的发生发展相关。
17 总结与展望
前列腺癌是男性最常见的癌症之一,其发生发展与AR 信号通路密切相关。然而,AR基因突变、基因扩增和剪接体异常表达使前列腺癌治疗面临耐药难题。因此,开发新靶点以克服临床耐药是该领域亟需解决的关键问题。
核受体AR 与其他核受体成员关联密切,科研人员期望通过对其他核受体进行研究以发现可用于前列腺癌治疗的潜在靶标。近些年研究发现,核受体LXRα/β,RORγ,RARγ,PPARγ,FXR,COUPTFII,TLX,TR4,ERα/β,ERRα/γ,LRH-1,SF-1,GCNF,SHP,DAX1,GR,PXR,VDR 和MR 等与前列腺癌的发生发展相关,同时已有越来越多天然产物或合成化合物作为核受体的激动剂或抑制剂用于抗前列腺癌的研究(见表1)。
总之,核受体在细胞的增殖、生长、存活和凋亡过程中发挥着重要作用,AR 以外的核受体有望成为前列腺癌治疗的新靶点。因此,探索核受体与前列腺癌的作用机制并寻找和开发抗前列腺癌的新核受体调节剂具有重要的研究意义和潜在的临床应用价值。
