金属氧化物异质结的构建及在光催化CO2还原反应中应用的研究进展
2023-10-17李孟蝶王祖民于然波
李孟蝶, 王祖民,, 齐 健, 于然波
(1. 北京科技大学冶金与生态工程学院, 北京 100083;2. 中国科学院过程工程研究所生化工程国家重点实验室, 北京 100190)
随着工业的发展, 能源短缺、 全球变暖等问题已经引发广泛关注[1,2]. 据估计, 由于CO2的过度排放, 会导致80年后平均气温升高1.9 ℃, 将会为人类生存带来巨大危机. 科学家们提出并实施了各种新技术用于CO2捕获、 储存和转化. 其中, 将CO2转变成具有高附加值的化学产品和燃料, 可以解决CO2过度排放造成的资源浪费、 环境污染和温室效应等问题, 并有望实现从化石燃料经济向可持续的CO2经济转变. 尤其是将太阳光作为唯一的能量输入, 模拟自然界的光合作用, 通过光催化的手段促进CO2转化成CO, CH4, CH3OH 和HCOOH 等化学品, 将是颇具前景的绿色新技术[3]. 然而, 线性结构的CO2分子具有较强的化学惰性, CO2的转化过程需要大量的能量输入来打开C=O 键[4](其键能为750 kJ/mol), 故需要引入均相和多相催化剂来活化CO2分子, 从而加速这一缓慢的还原反应. 虽然均相催化具有较高的选择性, 但大多数均相体系存在成本高、 稳定性差、 催化剂分离困难等问题, 限制了其实际应用; 而在多相催化反应中, 催化剂具有可循环利用的优势, 在CO2催化转化中已占有越来越重要的地位. 近些年围绕CO2光还原方面的研究主要集中于金属[5]、 金属氧化物[6~8]、 硫化物[9]、 碳基材料[10,11]和金属有机骨架(MOF)[12]等多相催化剂, 并且已经取得很大的进展.
太阳通过光辐射的方式每年入射到地球表面的能量多达1.3×105TW, 但是, 通常情况下 CO2在200~800 nm波长的可见光或紫外线辐射下是化学惰性的. 因此, 需要通过光催化剂吸收太阳光能量而产生光电子, 并迁移到光催化剂表面将吸附态CO2还原. 整个过程如图1 所示, 主要分为光捕获和吸收、 光生电荷分离和迁移以及催化剂表面CO2还原反应等3个步骤. 当有足够能量的光子入射到半导体催化剂上时, 会产生具有足够能量的电子去跨越带隙(Eg), 使得处于基态的价带(VB)电子跃迁到导带(CB)上并产生光生电子-空穴对, 但光生电子-空穴对迁移到催化剂表面的同时, 也往往伴随着电子-空穴对的复合, 这大大降低了反应的催化活性. 因此, 为了抑制或减少电子-空穴对的复合, 一个非常有效的方法就是构建异质结构来促进电子-空穴对的分离. 由此产生的电子-空穴对会迁移到催化剂表面, 并与吸附的反应物作用, 空穴参与氧化反应被消耗, 同时剩余的质子和电子将CO2还原成一氧化碳(CO)或其它碳氢燃料. 其中, 如果空穴被牺牲剂(三乙醇胺、 乳酸、 三乙胺等)消耗, 不仅浪费光生空穴的能量, 而且极大增加了整个反应的成本.空穴将水氧化而产生氧气(O2), 则无需额外添加牺牲剂, 更加接近大自然的光合作用, 是更加绿色环保和经济的过程. 因此, 本文讨论的绝大部分CO2光还原体系都是水参与的气体CO2分子还原, 后文如未特殊说明则均为无牺牲剂的气固反应体系.
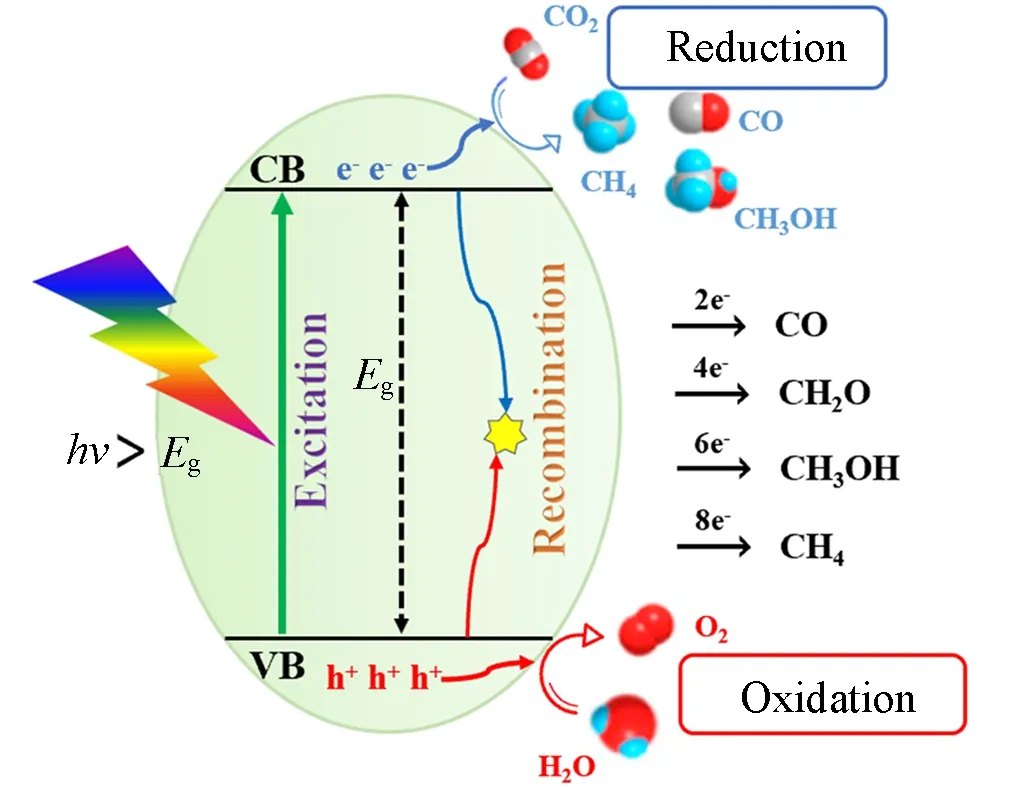
Fig.1 Schematic diagram of photocatalytic CO2 reduction
对于光吸收过程, 能带结构决定了材料对光的吸收能力. 通常, 带隙越窄的半导体能被更多波长的光所激发, 具有更宽的光谱吸收范围, 可以更加充分地利用光能. 然而对于表面氧化还原反应而言, 光生空穴和电子需要具有足够的氧化还原能力. 只有催化剂能带位置与CO2还原电势相匹配, 即光催化剂价带顶的位置应比H2O氧化产物的氧化电位更正, 使之被空穴氧化生成O2; 光催化剂导带底的位置应比CO2还原产物的还原电位更负, 热力学上才能发生还原反应(图2)[13]. 同时, 如果用水作为质子提供剂时也意味着催化剂的价带位置要正于水氧化电极电势, 氧化反应才能够进行[14~16]. 但这往往与上面讨论的带隙要求相违背, 难以在单一的半导体同时实现. 另一方面, 正如前文所述, 大多数传统的光催化剂(如TiO2, ZnIn2S4和g-C3N4)由于体相内部的光生电子-空穴极易复合, 表现出较差的光催化效率[17]. 因此急需优化光催化过程中的载流子输运, 抑制衰变路径, 才能实现CO2还原性能的显著提高. 而在动力学上, Zhao等[18]最新研究发现氧化物表面的CO2分子可以通过短暂捕获电子来激发分子的弯曲和非对称拉伸的振动模式, 降低其最低未占据分子轨道能量, 使得CO2分子能够捕获光电子, 发生还原反应, 因此同样需要延长捕获电子的寿命. 如何充分利用光能, 产生尽可能多的光生载流子, 并有效提高载流子的分离效率, 是制备高性能光催化CO2还原催化剂的关键问题[19,20].

Fig.2 Reduction potentials of different products and corresponding reduction/oxidation potentials of several photocatalysts[13]
金属氧化物作为一类丰富廉价、 稳定安全和应用最为广泛的半导体材料, 其光催化剂的性能主要取决于其能带结构设计[21,22]. 传统上, 通过添加金属阳离子降低导带底, 或掺杂非金属离子(如C, N,S, F)提高价带顶, 从而减小半导体带隙, 以拓展半导体光的吸收范围. 如, 通过Pt2+掺杂可有效将TiO2光吸收从紫外光区拓宽到可见光区[23]. 此外, 也常常通过沉积贵金属(如Pt, Ag, Pd, Au)来提高光催化活性. 在金属复合体系中, 光生电子聚集在金属上, 而空穴则留在半导体表面, 能够有效抑制载流子的复合. 更重要地, 大量研究表明, 在两个电子带结构趋同的半导体之间形成异质结, 也可有效抑制光生电子和空穴的复合. 因此, 构建具有优化能带结构的氧化物半导体异质结具有诸多优势. 一方面, 适当的异质结构可调节其能带结构, 使其充分利用光能, 增加产生的光生载流子. 另一方面, 异质结的界面处会产生内建电场, 促进其在异质界面上转移以抑制复合, 进而增加光生电子的利用率.此外, 由于异质结的表面和界面的性质深刻影响着催化剂对CO2的吸附形式和强度, 这对光催化CO2还原的选择性起着至关重要的作用. 如共催化剂引入的额外活性位点有利于CO2的吸附和活化, 从而提高CO2的光还原效率. 因此构建异质结有望在提高量子效率的同时也能提高产物选择性(图3)[24].
1 异质结的构型
由不同组分复合形成的异质结构, 具有实现电荷载流子在界面上空间分离的潜力, 可以抑制或减少电子空穴复合, 延长载流子寿命. 异质结可以在金属和半导体之间以及具有不同间隙的半导体之间形成. 因为载流子的运动必须严格按照电势梯度进行, 而不同材料中的内建电场、 载流子浓度和迁移率不同, 所以异质结的种类对其催化性能具有显著影响.
在最常见的金属/半导体异质结中, 因功函数(φ)的差异以及费米能级(EF)的相对位置不同, 金属与半导体在界面处可能形成肖特基势垒或欧姆接触. 当金属和半导体接触时, 自由电子将由功函数较低的材料流向功函数较高的材料, 直到达到平衡, 并在界面处建立亥姆霍兹双电层, 称为空间电荷层. 以在n型半导体(电子为多数载流子)为例, 当金属的功函数(φm)>半导体的功函数(φs)时(如图4左侧所示), 半导体的电荷将流向金属, 费米能级降低, 导致其表面附近的载流子浓度相比于本体减少,即形成电子耗尽层, 故半导体保留过量正电荷. 相反, 如图4右侧所示, 当φm<φs时, 由于电子从金属转移到半导体, 则产生电子累积层. 同时, 由于电荷转移, 半导体和金属之间产生电场, 半导体中的能带边缘也连续移动, 当φm>φs时, 能带在界面处向上弯曲; 相反φm<φs时, 能带向下弯曲. 而界面处半导体能带的弯曲程度取决于金属和半导体之间的功函数差. 所以, 当n型半导体中φm>φs时, 在金属-半导体界面处会形成一个肖特基势垒, 半导体产生的光生电子会自发地转移到金属表面, 而空穴保留在半导体内部, 从而实现了电子和空穴在空间上的分离, 提升载流子的寿命. 而φm<φs时, 则不能形成肖特基势垒, 只能产生欧姆接触[25].
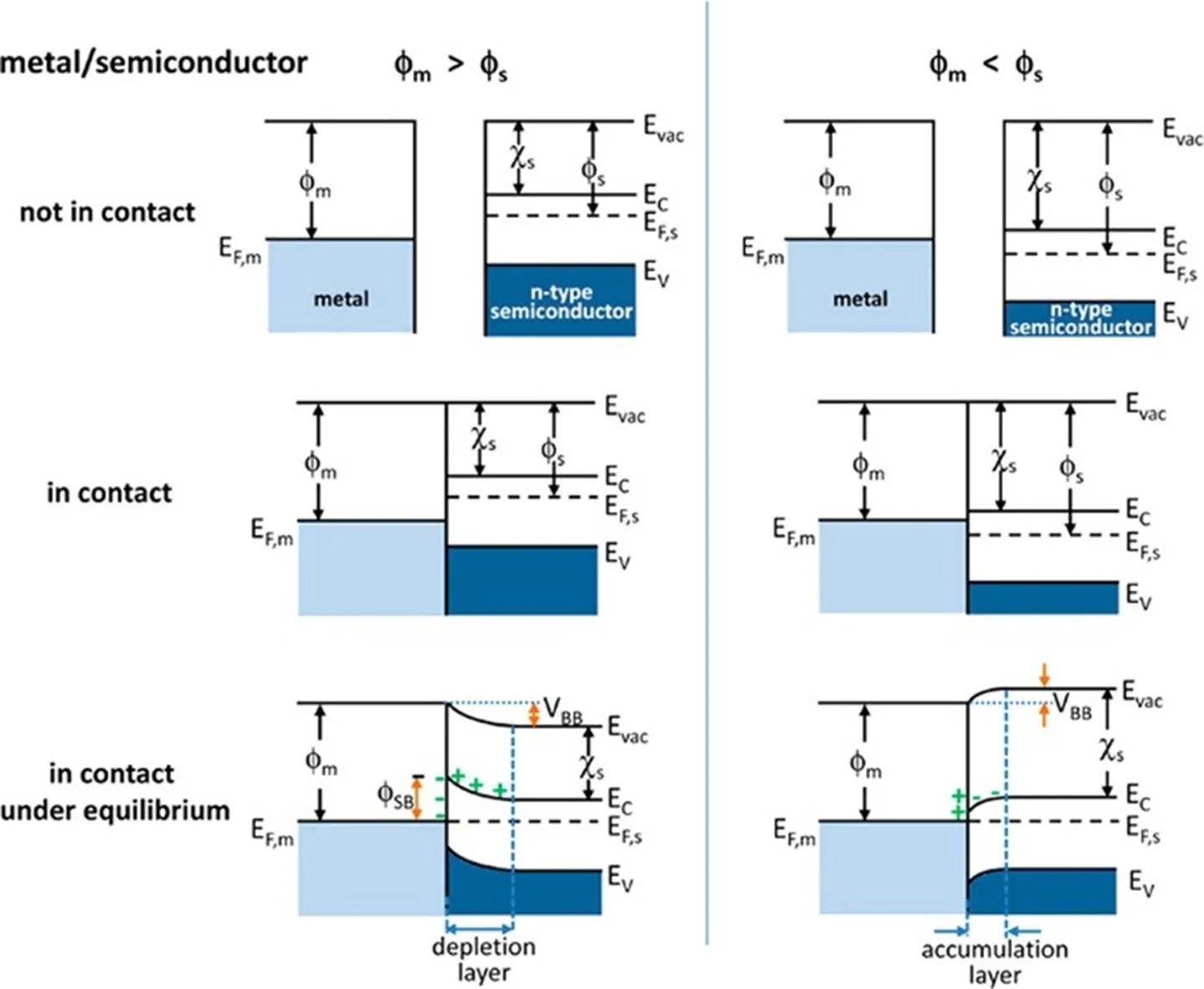
Fig.4 Energy band diagrams of metal and n-type semiconductor contacts[25]
类似地, 从半导体中提取载流子以降低电荷载流子复合的另一种常用策略是与其它半导体复合,即构筑由两个或多个具有不同能带的半导体材料紧密结合的半导体/半导体异质结. 根据半导体的VB和CB 电位和禁带宽度, 复合材料的电子结构可分为Ⅰ(跨隙)、 Ⅱ(错隙)和Ⅲ(断隙)型3 种异质结[图5(A)~(C)][26]. 在I型构型中[图5(A)], 窄带隙半导体(半导体2)的VB和CB电位被限制在宽带隙半导体(半导体1)内部, 形成一个跨带排列. 因此, 当足够能量的光照射时, 两个半导体中均产生光生电子-空穴对, 而所有载流子移动并聚集在异质结构的一个组分上[27]. 由于电荷载流子在窄带隙半导体中积累, 电荷分离没有明显的整体增强, 光生电子空穴的能量却显著降低, 往往反而会降低光催化效率. 在Ⅱ型异质结中[图5(B)], 半导体1 和2 之间的带隙电位是交错的. 由于两个半导体之间的电势差, 形成了一个上升或下降的带状弯曲, 这迫使电荷载流子向相反方向迁移. 这大大改善了异质结各部分的电子-空穴空间分离, 以延缓电荷重组, 延长自由电子和空穴的寿命. 同时, 氧化和还原反应发生在两个不同的半导体表面, 避免了逆反应的发生. 本文中描述的大多数异质结复合结构都属于第Ⅱ型排列. 另外, 在III型异质结中[图5(C)], 一个半导体(半导体2)的VB和CB边缘均高于半导体1的CB 电位, 没有重叠, 两个半导体之间没有电荷迁移, 因此不能促进有效的电荷分离. 基于第二类排列, 虽然可以实现有效的电荷分离, 但其主要的缺点是在电荷转移过程中, 所有电子均转移到具有较小的CB负电位的半导体2上, 而所有空穴则均转移到具有较低的VB正电位的半导体1上, 电荷载流子的氧化和还原能力降低. 最近, 人们还发现了一种异于传统Ⅱ型错隙结构的新型异质结, 即所谓的Z型异质结[图5(D)], 其可以克服上述不足. 在Z型异质结界面处形成的内建电场中, 具有较低导带位置的半导体受光激发产生的电子可以与具有较高价带位置的半导体中空穴结合. 而各个半导体中剩余的电子和空穴仍保留在相应的更负的CB边缘和更正的VB边缘, 将具有更高的光催化氧化还原电位. 因此, 这种独特的异质结构在实现高效的电荷分离和强氧化还原能力上具有双重优势[28].
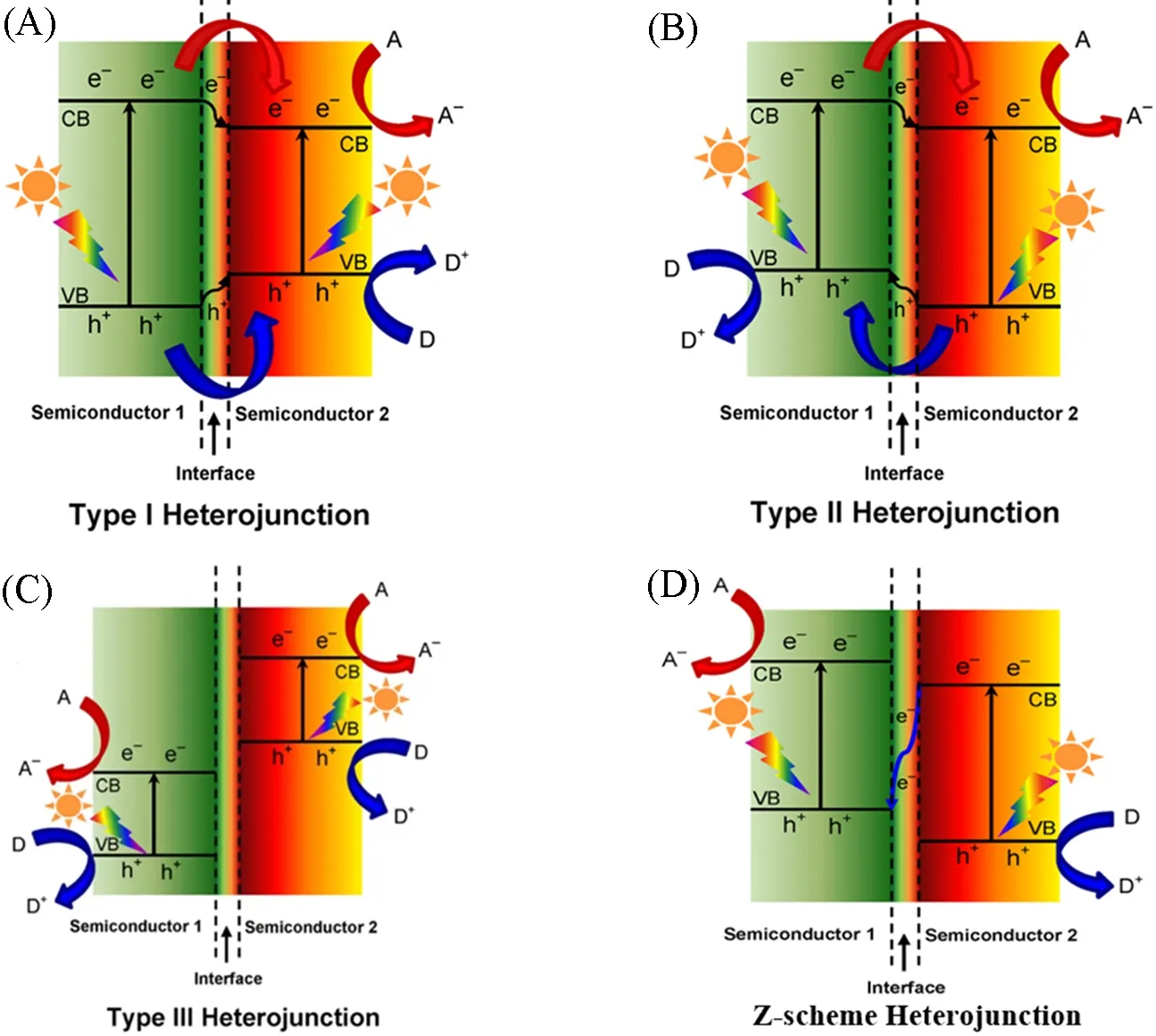
Fig.5 Schematic energy band diagrams of three different types of heterojunctions of type I(A), type II(B),type III(C) and Z-scheme(D) in a typical semiconductor hybrid nanocomposite[26]
2 氧化物异质结构
过渡金属氧化物(如TiO2, ZnO, CuO和Fe2O3等)因其独特的性能、 高稳定性和低成本等优势, 在多相催化中得到了广泛应用. 以TiO2为主要代表的宽带隙金属氧化物难以对可见光响应, 如锐钛矿型TiO2只在小于380 nm的紫外光下受到光激发. 而具有窄带隙的氧化物[如WO3(带隙2.4~2.8 eV)]可以在较大范围响应太阳光, 但由于导带电位较低其光生电子的还原能力不足, 导致其CO2还原性能不佳[29]. 因此, 需要将半导体氧化物与不同材料进行复合, 构建多种不同种类的氧化物异质结构, 以提高光响应范围, 促进电荷分离, 优化反应物和中间产物吸附活化, 提高光催化CO2的活性和选择性.
2.1 氧化物/金属异质结
与块体金属相比, 金属纳米颗粒(NP)由于其较大的比表面积、 丰富的活性位点(不饱和配体原子)和独特的电子结构, 在催化应用方面引起越来越多的关注[30]. 然而, 由于金属纳米颗粒具有高表面能, 在热力学上表现非常不稳定, 所以其在催化反应中很容易发生迁移和团聚. 这种结构变化通常伴随着催化活性和选择性的快速下降. 因此, 将金属纳米颗粒与载体复合可以在保持表面暴露的情况下, 避免颗粒的聚集, 从而提高其催化稳定性[31].
在金属-氧化物复合催化剂体系中, 催化剂的活性位不仅可以是金属, 也可以是氧化物和金属-氧化物界面. 一般来说, 对催化作用的影响源于金属纳米颗粒和氧化物载体之间的相互作用[32]. 这些相互作用包括金属和氧化物间的电子转移、 界面附近反应物种的迁移、 氧化物对金属纳米颗粒形貌的调控、 氧化物对金属纳米颗粒化学成分的影响以及氧化物向金属表面的电子转移(强金属-载体相互作用, SMSI)(图6)[32]. 为了调节金属-氧化物之间的相互作用, 可以从金属纳米颗粒的类型、 大小、 形貌和结构等方面进行调控.
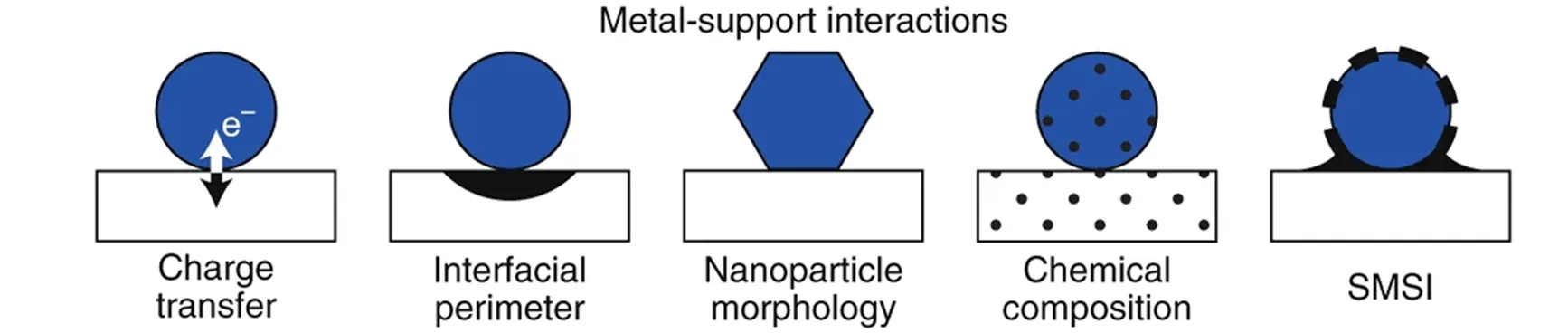
Fig.6 Interactions present in the metal-oxide composite nanocatalyst system[32]
一般而言, 负载的金属会与半导体之间形成肖特基势垒, 捕获电子, 避免其与空穴复合. Zhang等[33]将Ag纳米颗粒(Ag NPs)负载在Cu2O上, 并与ZnO之间形成Z型异质结. 研究发现, Ag NPs助催化剂加速了电子从半导体的抽离, 并作为“电子蓄水池”在其表面富集大量电子, 电子与吸附的分子发生快速电荷转移, 进行催化反应, 提高了CO2光还原活性. 同时, 由于Ag 捕获Cu2O 表面积累的过量电子, 这可以缓解Cu2O的自身光还原, 进一步提高Ag-Cu2O/ZnO NRs的光催化稳定性[图7(A)]. 光催化将CO2转化为HCOOH是解决能源供应长期需求的一个备受期待的方法. Wang等[34]通过对Cu-MOF前驱体的热处理, 制造了由Cu核心包裹Cu2O外壳的均匀的核壳纳米粒子, 用于H2O存在下液固相光催化CO2还原. Cu@Cu2O/C-350在CO2还原成HCOOH的过程中表现出很好的催化活性, 在室温下不需要任何添加剂就能达到31 μg/h的HCOOH生产速率, 由于Cu@Cu2O/C-350中金属铜芯的表面等离子体共振(SPR), 560 nm处的表观量子效率(AQE)为0.12%. 进一步的机理研究表明, 表面的Cu2O外壳作为活性点吸附并活化了CO2[图7(B)].
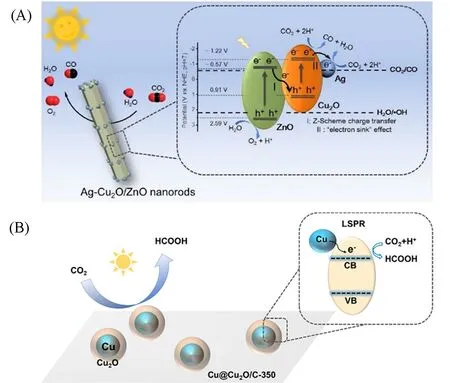
Fig.7 Possible mechanism of Ag-Cu2O/ZnO NRs photocatalyst in CO2 reduction process(A)[33] and possible mechanism of Cu@Cu2O/C-350 photocatalyst in CO2 reduction process(B)[34]
金属纳米颗粒的种类是影响催化作用的关键因素. 一般来说, 金属助催化剂(Pt, Pd, Rh, Au, Ag等)的负载可以提高还原反应光生电子的消耗速率, 加速表面还原反应的进行. 为了进一步了解金属粒子助催化剂的作用, Xie等[35]在气-固界面反应下研究了不同贵金属助催化剂对CO2光催化还原的影响规律, 研究发现, 电子消耗速率R(e-)呈TiO2<Ag-TiO2<Rh-TiO2<Au-TiO2<Pd-TiO2<Pt-TiO2的顺序增加(表1). 这一趋势与贵金属功函数的变化(Ag<Rh<Au<Pd<Pt)增加趋势相对应. 由于功函数反映了电子的提供能量或接受能量的能力, 因此他们认为, 金属作为助催化剂可能会将TiO2中的电子提取出来,从而抑制光生电子-空穴对的复合. 瞬态光电流测试的结果也证实了这一猜想.
影响催化作用的另一个关键因素是金属纳米颗粒的尺寸. 一般来说, 金属与半导体间的相互作用(特别是电子转移)在金属纳米粒子尺寸降低后会变得更加显著. Ma等[36]为了更好地理解氧化物负载贵金属在CO2还原过程中的作用, 研究了负载金属粒径对Pt/ZnO 催化剂性能的影响. 随着Pt 负载量的增加, Pt 纳米颗粒的尺寸也在增长[图8(A)]. 其性能如图8(B)所示, ZnO-0.75Pt 异质结纳米复合材料在300 W 氙灯照射下, 显示出比纯ZnO 纳米纤维优越的光催化活性, CO 生成速率高达45.76 μmol·g-1·h-1. 由图8(C)可见, 在ZnO纳米纤维的表面引入合适粒径的Pt纳米颗粒, 能显著扩大光的吸收和利用. 此外, Pt增加了额外的反应活性位点, 并促进光产生的电子-空穴对的分离, 表明载流子寿命得到延长[图8(D)和(E)]. 异质结结构结合纳米纤维在潮湿的反应环境中电荷载流子传输路径短和自支撑效应的天然优势, 显著提升了光催化性能.
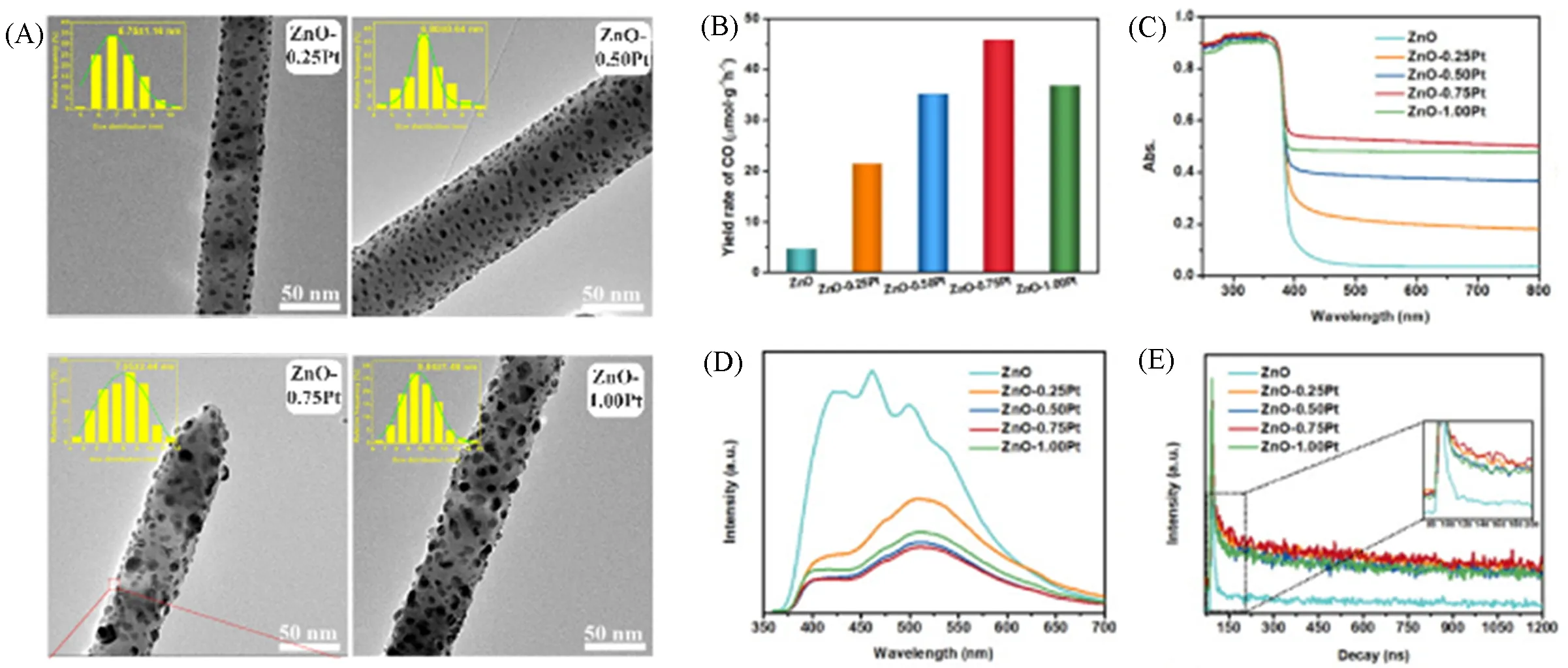
Fig.8 TEM images of Pt/ZnO with different Pt loadings(A), comparison of CO yields of bare ZnO fibers and Pt/ZnO nanocomposites(B), UV-Vis spectra(C), steady-state(D) and time-resolved PL spectra(E)[36]
此外, 近20 年来, 一直有研究利用Au 和Ag 纳米粒子及其强大的SPR 来提高光催化效率. 如Xue等[37]成功制备出了一种新型的金纳米粒子修饰的有序介孔二氧化钛(OMT)复合材料, 均匀分散在TiO2上的Au NPs 受到光激发, 由于SPR 效应, 产生大量热电子, 并快速稳定地传输到TiO2表面, 使得该TiO2催化材料在可见光下和H2O存在下的液固相光催化显示出很高的光催化CO2还原性能. 同时有序介孔TiO2的高表面积提供了更多的反应位点, 三维传输通道保证了气体分子的顺利流动和高效的CO2吸附, 这使得光催化性能进一步提高(图9). Li等[38]研究了Ag 纳米粒子修饰的TiO(2Ag/TiO2)异质结构对CO2的光还原, 由于Ag NPs 的SPR 效应扩展了对可见光的捕获和利用, 并加速了表面还原反应, 催化剂显示出了优异和稳定的光催化活性.
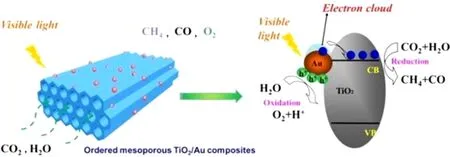
Fig.9 Possible mechanism of the OMT-Au photocatalyst during CO2 reduction[37]
表2 列出了最近利用Au NPs, Ag NPs, Cu NPs 表面等离子共振效应开发的多种CO2光催化剂[34,39—43].
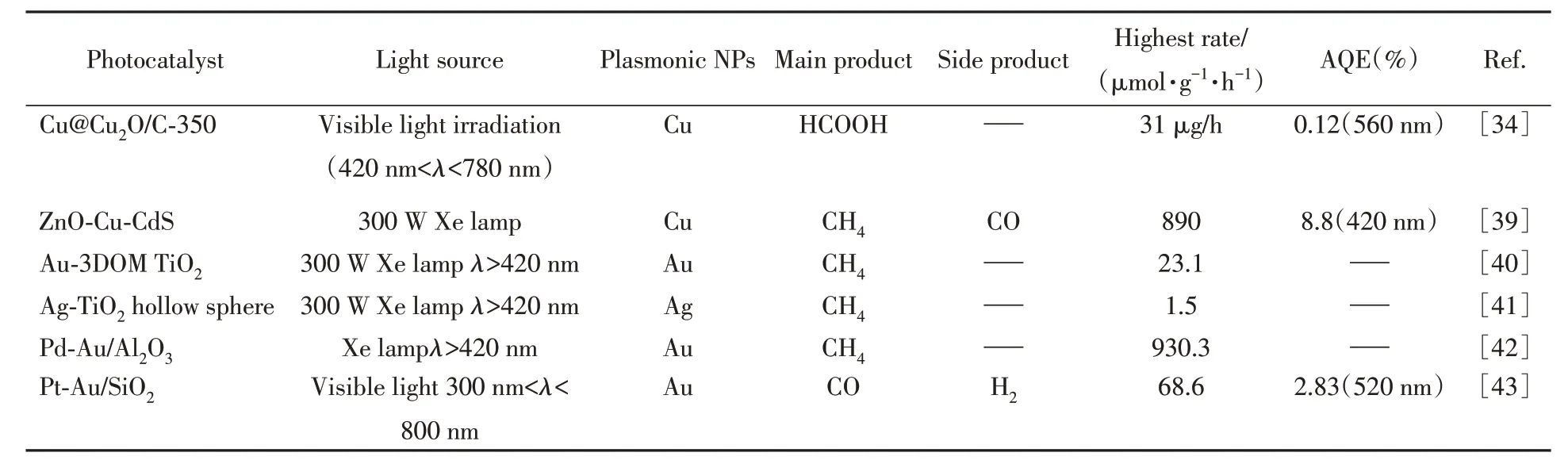
Table 2 Photocatalysts developed using the SPR effect of Au NPs, Ag NPs
2.2 氧化物/氧化物异质结
针对单一氧化物, 当CB不够负、 VB不够正时没有足够的氧化还原能力, 或带隙Eg过宽而不能有效吸收光, 其能带结构均难以满足高效CO2光还原的要求. 而将多种氧化物复合, 形成异质结构, 优化能带结构, 可以取长补短, 产生更多有效的光生电子-空穴对, 提高CO2光催化活性. 如, TiO2由于其宽带隙, 只对紫外光有响应, 使其对太阳光利用率低, 导致光催化效率较低. 为了提高TiO2纳米纤维在可见光区的吸收, 将CuO负载在TiO2纳米纤维上构建异质结构[44]. n型TiO2半导体的费米能级Ef,n接近导带区, p型CuO半导体的费米能级Ef,p接近价带区. 二者结合后, 不同的费米能级会导致电子从TiO2转移到CuO上, 这会使CuO费米能级上升, TiO2费米能级下降. 此时, n-TiO2与p-CuO之间会形成内建电场. 在可见光照射条件下, CuO导带上的激发电子在内建电场的驱动下, 会转移到TiO2导带上, 而空穴留在CuO 价带上. 由于能级匹配良好, 构建了II 型能带对齐结构, 促进了p-n 异质结中加速电荷转移. 结果表明, TiO2负载CuO 后对可见光的吸收增强, 荧光强度下降, 光生电子-空穴对的复合率降低, 光催化性能得到显著提升. 在加入了还原剂乙二胺的液固体系下, 在300 W氙灯照射4 h, 催化CO2和H2O反应中, CuO/TiO2纳米纤维催化的甲醇产率为1674 μmol/g, 较纯TiO2纤维提高了4.4倍.
通过形成异质结结构, 还可以提高还原产物的选择性, Nogueira等[45]发现CuO纳米颗粒与Nb2O5结合, 可以提高其在H2O 存在下液固相光催化CO2还原性能. 形成的异质结有效增加电荷分离效率, 但更重要的是影响副产物的选择性, 在低CuO含量(2.5%, 质量分数)时, CO2的还原效率提高, 导致液态烃(HCOOH和CH3COOH)的形成; 而在较高含量(10%, 质量分数)时, CH4的生成率提高. Nb2O5和CuO表面可能吸附CO2的行为如图10所示, 虽然CO2是一个没有偶极矩的线性分子, 但两个O原子都有一对可以转移的电子, 所以它可以表现为路易斯碱; 另一方面, C原子的行为类似于路易斯酸并接收一对电子. 根据Nakajima 等[46]的报道, NbO4四面体存在于Nb2O5结构中, 在水存在时形成NbO4-H2O, 水的作用是由于路易斯酸位点从CO2的氧中接受一对电子, 因此来自CO2的氧与Nb2O5表面形成主要配位,而CuO纳米颗粒的表面通过向C原子提供一对电子而充当路易斯碱中心. 当CuO在Nb2O5表面充分覆盖时候, CO2大多数通过C原子与路易斯碱中心配位的模式吸附, 可以充分脱除O原子, 并逐步与多个H原子结合, 因此可以高选择性地深度还原为CH4.
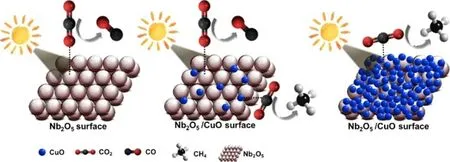
Fig.10 Scheme of possible CO2 adsorption on the surface of Nb2O5 and CuO[45]
利用太阳能将二氧化碳还原反应(CO2RR)转化为有价值的碳氢化合物为低碳未来提供了有吸引力的解决方案, 但其性能受到同时发生的竞争性析氢反应(HER)的影响. 为此, Dou等[47]提出了最大化界面集成和表面重构工程策略, 以提高异质结光催化剂的CO2RR活性和选择性. 表面重构ZnO/CuOx催化剂均匀地锚定在由碳纳米纤维支撑的多孔碳纳米片阵列上(ZnO/CuOx-C CNFs). 通过减小ZnO/CuOx的尺寸可以最大限度地实现各组分的界面整合, 促进电子-空穴对分离, 增加表面活性位密度. 此外,通过简单的光照射在ZnO上形成羟基的表面重构过程, 促进了CO2RR生成CH4和产氧反应(OER)的动力学, 抑制了H2和CO的竞争生成(图11). 这些优点使ZnO/CuOx-C CNFs具有优异的催化性能: 在全光照射下, CH4的生成速率可达241.6 μmol·g-1·h-1, 选择性可达96%.
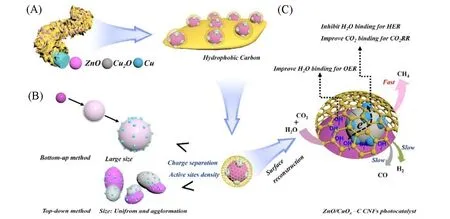
Fig.11 Hierarchical structure of ZnO/CuOx heterojunction embedded in porous carbon nanosheet array supported by CNFs(A), the particle size of ZnO/CuOx smaller than previously reported heterojunction photocatalysts synthesized by top-down and bottom-up methods(B), the surface reconstruction induced by solar light irradiation leading to rich accumulated electrons for CO2RR towards CH4, and inhibiting the H2 and CO generation(C)[47]
通过选择合适的氧化物材料形成异质结, 可以有效提升CO2光还原活性和选择性, 异质结构中不同材料的含量同样对CO2光还原性能有着显著影响. Matejová 等[48]通过溶胶-凝胶法合成了Ce 含量为0.28%~10%(摩尔分数)的TiO2-CeO2复合材料, 并测试了这些材料在紫外线照射下以0.2 mol/L NaOH作为还原介质的液固相光催化CO2还原性能. 他们发现, Ce 的引入不仅可以将Ce4+掺入锐钛矿晶格,使吸收光谱由紫外光向可见光区域转移, 而且负载的CeO2数量对材料导带和价带位置有着关键影响.由于功函数和带隙能量对TiO2中Ce负载的依赖性, 导带电子的能量受到CeO2添加的显著影响, 而这直接影响了CO2的光还原过程[图12(A)]. 当低浓度负载时, 电子和空穴都具有光催化CO2所需的还原和氧化电势. 当TiO2复合材料中的Ce 含量较高时(≥3%, 摩尔分数), CB 中电子的能量下降到所需的H+还原电位以下, 因此这些催化剂在CO2光还原中的性能显著下降[图12(B)和(C)].
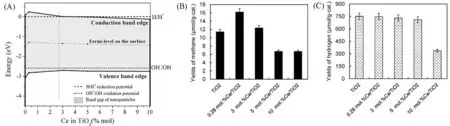
Fig.12 Illustration of the shifts of energies of electrons and holes in conduction and valence bands in dependence on the Ce loading(A), yields of methane(B) and hydrogen(C) over individual investigated photocatalysts in the CO2 photocatalytic reduction[48]
2.3 氧化物/硫化物异质结
金属硫化物的导带通常由d轨道构成, 而价带由S3p轨道构成. 由于金属硫化物的3p轨道比相对应的氧化物的O2p轨道能量要高, 通常其价带位置更负, 带隙宽度更窄[49], 能够吸收更大范围的太阳光,具有更强的光电子还原能力. 因此, 金属硫化物通常比对应的氧化物有更好的光催化性能[50,51]. 但是硫化物半导体在液相中的稳定性表现得较差, 晶格中的S2-易被光生空穴氧化(S2-+2h+→S), 在一定程度上限制了金属硫化物的实际应用. 相比之下, 金属氧化物的化学性质稳定, 并且具有和硫化物互补的正价带位置. 因此, 通过构建异质结构能够充分利用各自的优势, 不仅可以拓展光催化剂的光吸收范围, 而且还可以促进电荷载流子的分离. You等[52]通过控制材料组成的部分晶体结构, 构建了具有局域晶格畸变的SnS2/SnO2异质结构. 位于SnS2/SnO2纳米界面的晶格畸变可以提供额外的活性位点,不仅在可见光下具有催化活性, 而且还可以改善光激发电子-空穴对的分离. 并引入具有高效光捕获能力的中空多壳层结构(HoMSs), 进一步提升光催化效率. 催化剂在气-固体系中实现了较高的CO2还原催化活性(48.01 μmol·g-1·h-1), 并且具有优异的稳定性和100%的CO选择性(图13). 同时提高硫化物在氧化物表面的分散度对于提升CO2的光还原具有重要意义. Li等[53]通过简单水热法和连续离子层吸附反应, 将CdS量子点均匀分布在WO3纳米片表面形成异质结. WO3纳米片可以极大地提高CdS量子点的分散性, 从而提高CO2在光还原位点的吸附和反应效率, 在以三乙醇胺为牺牲剂的条件下, 其CO和CH4的生成速率分别可达64.7和2.3 μmol·g-1·h-1.
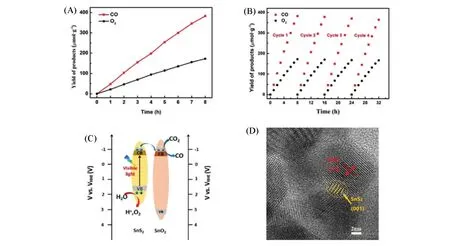
Fig.13 Yield(A) and stability(B) of CO2 photoreduction of CO and O2 under visible light irradiation,diagram of possible energy band structure and main CO2 reduction mechanism(C) and highresolution transmission electron microscopy image(D)[52]
同时, 硫化物与对应的氧化物紧密结合, 由于较大的晶格参数差异, 往往会在界面处形成内电场(IEF). 这种内建电场在加速电荷分离和迁移方面展现出独特的优势[54~56]. Chen 等[57]发现从原始的二维(2D)金属硫化物构建氧化物/硫化物平面异质结可以有效地增强IEF 强度. 以二维In2S3纳米片为模型, 通过O2等离子体技术获得In2O3/In2S3平面内异质结, 其IEF 显著提高2.7 倍. 得益于IEF 的增大,In2O3/In2S3的电荷分离效率提高了6.2倍, 达到5.6%, 同时电荷迁移速率也大大加快. 因此, 在三乙醇胺存在下的液固相CO2光还原过程中, CH4的生成速率提高了6.4倍(图14).
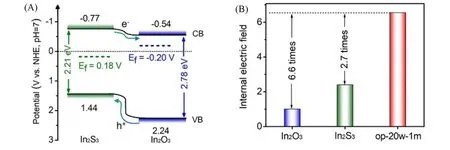
Fig.14 Band gap structure and charge migration(A) and internal electric field strength(B) of In2O3/In2S3 in-plane heterojunction[57]
2.4 氧化物/磷化物异质结
金属磷化物根据其金属种类、 金属磷比(M/P)和晶体结构表现出不同的特性[58,59]. 1988 年,Schnering等[60]报道了大多数富金属磷化物具有与金属和陶瓷相当的特性. 它们不仅具有优良的导热性和导电性, 而且具有良好的热和化学稳定性. 近年来, 分子式为MxPy的过渡金属磷酸盐(TMP)被用作催化剂. 由于P 的电负性(2.1)比大多数金属都略高, 因此P 与金属的结合限制了金属中的电子离域, 导致电子从金属转移到P. M—P 键的性质取决于不同金属的电负性差异和TMP 中的M/P 比率[图15(A)和(B)][61]. 此外, 随着M/P比的变化, TMP具有多种不同的晶体结构[图15(C)][62]. Diplas和Løvvik[63]指出Ni 和P之间的d电子态密度与Pt 的d电子态密度相似, 这意味着Ni2P在某些情况下可以表现出与贵金属类似的行为.
因此, 研究者开始了关于过渡金属磷化物用于光催化CO2还原的研究. 如Xu等[64]发现金属磷化物作为一类将高度分散的金属纳米团簇整合于P晶格中的固体, 可以提供理想的原子分布和晶体结构.因此他们选择Ni12P5为一个典型的磷化物, 研究其在整个太阳光谱范围内催化CO2还原的性能. 由于Ni12P5具有全光谱吸收和独特的线性吸附位点, Ni12P5可作为水汽逆反应的光热催化剂, 10.4% Ni12P5/SiO2的CO 生成速率为(960±12) mmol·g-1·h-1, 选择性接近100%, 长期稳定性好(表3). 他们将该研究成功地推广到Co2P, 同样获得了良好的CO2光催化性能. 这意味着金属磷化物可以作为高活性和高选择性的光热CO2催化的通用平台. Marci等[65]通过构建GaP/TiO2异质结催化体系, 可用于H2O存在下气固相光催化CO2还原, 通过10 h 性能测试发现, 对于纯相GaP, CH4的产率几乎可以忽略不计, 而GaP/TiO2(质量比1∶10)掺杂之后, CH4产率可达118.18 μmol/g. 他们发现由于TiO2和GaP 中CB 和VB的相对位置差异, 紫外光激发的TiO2导带中的电子可以被可见光激发的GaP 价带中的空穴捕获(图16). 因此, 二者的耦合机制不仅能有效地将TiO2的VB 中的空穴与GaP 的CB 中的电子分离利用,还可以实现光氧化还原过程的协同, 同时完成H2O的氧化和CO2的还原.

Table 3 Summary of the properties and catalytic performance of representative Ni12P5 samples
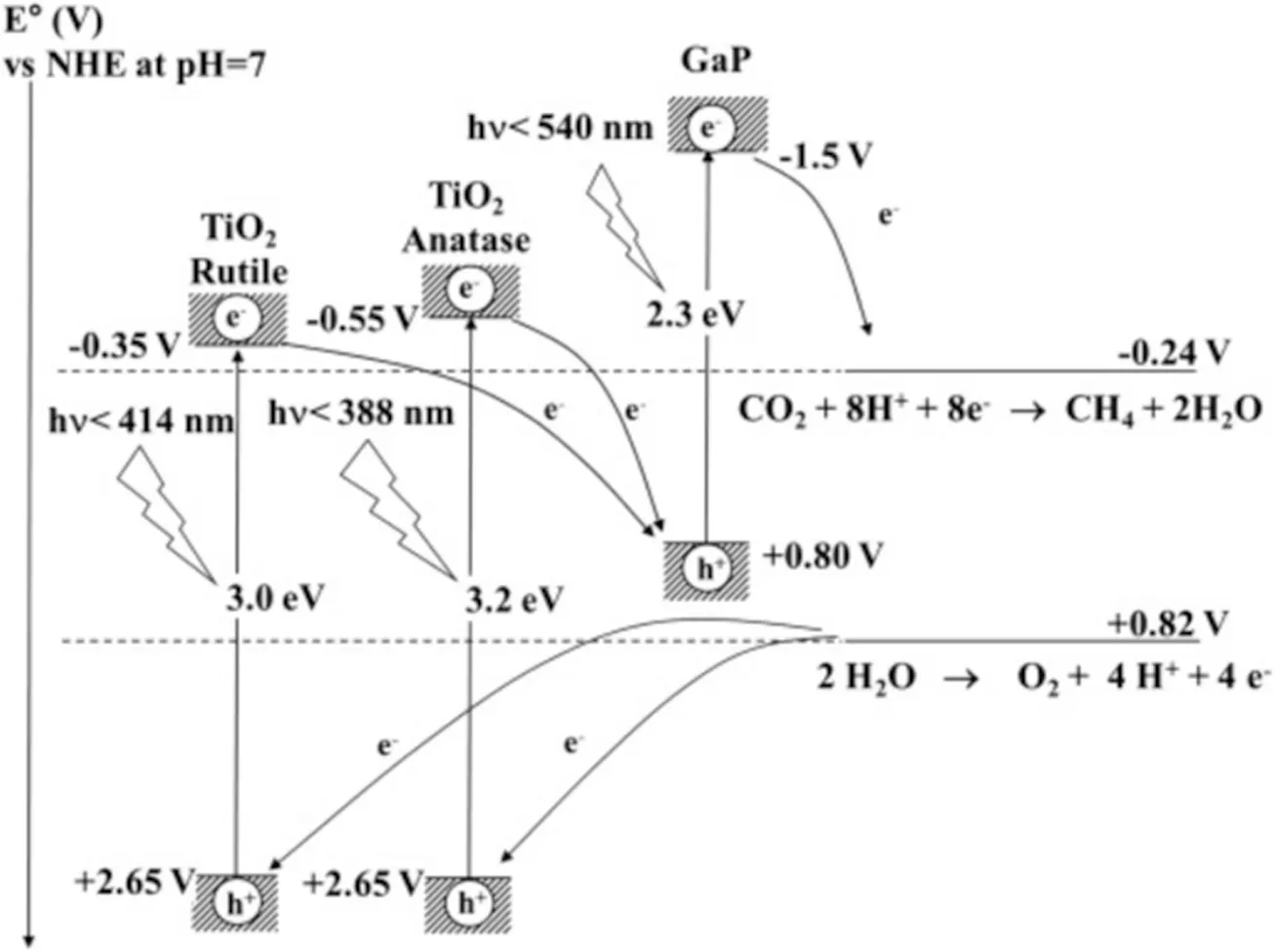
Fig.16 Relationship between the band structures of TiO2 and GaP and the reduction potentials(versus NHE at pH=7) for the most favorite processes of oxidation and reduction[65]
因此, 参考传统的贵金属/氧化物异质结的思路, 将贵金属替换为TMP, 不仅可以降低催化剂成本, 还有望通过调控TMP的电子结构, 调控CO2的吸附强度和吸附方式, 实现活性和选择性的进一步提升. Wang等[66]通过一个管状的氧化铟@磷化铟/氧化亚铜(In2O3@InP/Cu2O)的光催化剂来实现高效的光催化CO2还原, 该催化剂是由In-MIL-68 退火磷化后电沉积Cu2O 制备所得. In2O3的部分磷化使In2O3@InP/Cu2O具有强的光捕获能力. In2O3@InP异质结与原位负载的Cu2O相结合, 提供了一个双Z型异质结, 实现了高效的电子-空穴对分离, 其机理如图17 所示. 在各成分的协同作用下, 优化后的In2O3@InP60/Cu2O-1表现出对CO2的强吸附和高效的还原性能. 在模拟太阳光照射下, 加入了KHCO3的气固体系5 h后, CO和CH4的产率分别为13.7和38.8 μmol/g, 高于In2O3的相应产率(CO: 7.2 μmol/g,CH4: 13.2 μmol/g).
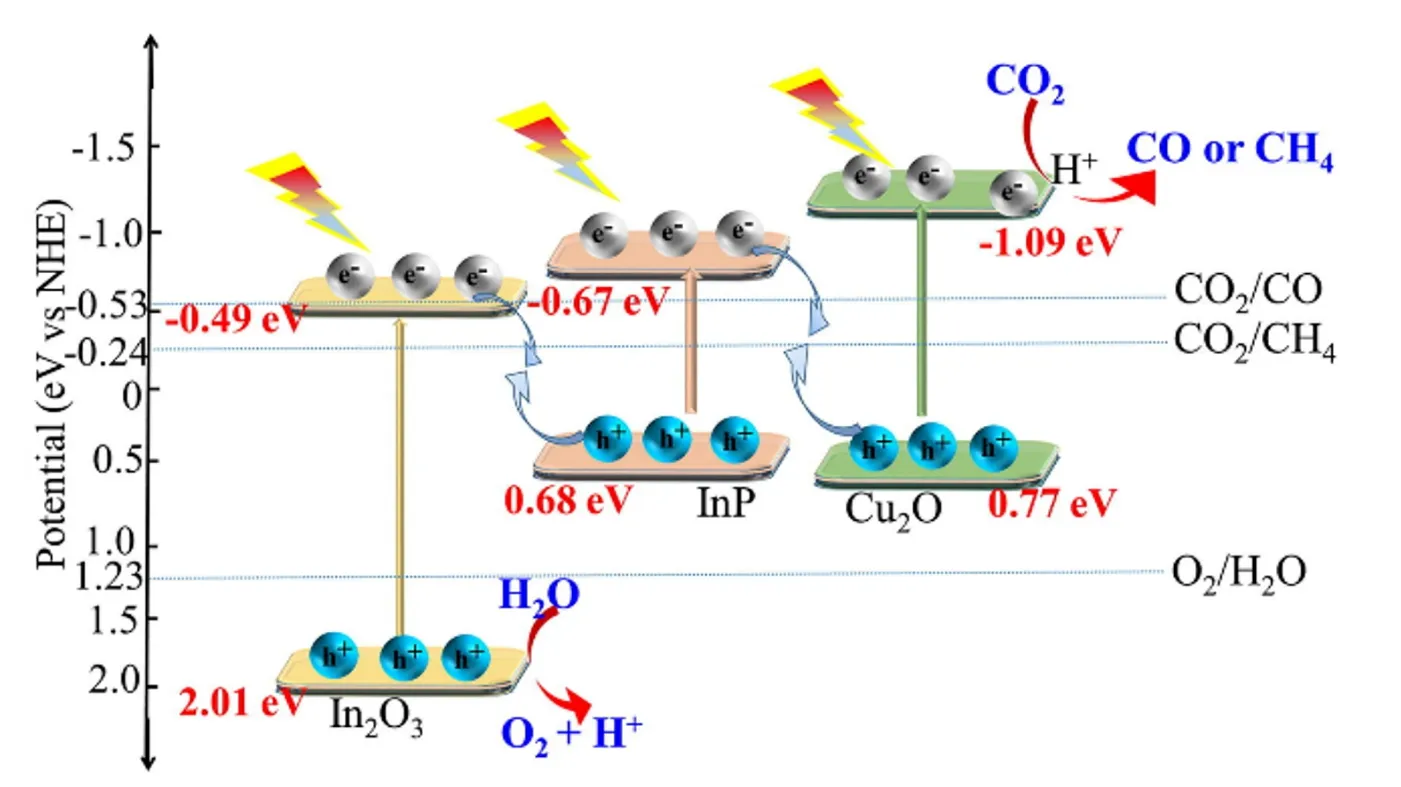
Fig.17 Double Z-type charge transfer mode and proposed mechanism of photocatalytic CO2 reduction with water as reducing agent[66]
综上分析, 在氧化物上负载非贵金属磷化物可以显著提高CO2还原性能(表4)[65~67], 该领域虽然处于起步阶段, 相关研究较少, 但是TMP/MO体系在成本控制、 活性增强、 选择性提高等方面的优势已经体现, 将会是一个值得研究的重要方向.

Table 4 Typical photocatalytic CO2 reduction systems using transition metal phosphides as co-catalysts
2.5 氧化物/氮化物异质结
金属氮化物各方面性质类似于金属, 即硬度高、 熔点高、 化学稳定性优异、 导电性好. 形成的金属-氮键促使氮化物产生缺陷, 具有给电子特性, 可提升光催化活性[68]. 因此, 研究者们开始将金属氮化物材料用于光催化CO2还原. Nguyen等[69]开发了一种基于一维TiN纳米管周期性阵列的三元异质结构催化剂[图18(A)~(C)], 该催化剂具有TiO2纳米中间层和In2O3-x(OH)y纳米壳[图18(D)]. 光照射到TiO2纳米颗粒层表面产生光生电子和空穴, 而后这些电子和空穴被转移到In2O3-x(OH)y纳米颗粒上的活性位点上, 并在此参与催化剂表面反应. 同时, 由于TiN纳米管具有很强的吸光性, 不仅为光催化反应提供了光热驱动力, 而且还可以作为一个关键的支架, 能够最大限度地暴露In2O3-x(OH)y纳米颗粒的活性位点, 从而提高CO的生成速率[图18(E)].
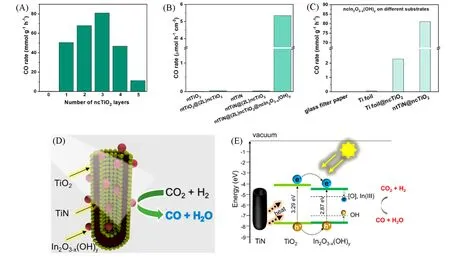
Fig.18 CO production rates in the reactor for ntTiN@ncTiO2@ncIn2O3-x(OH)y with different numbers of ncTiO2 layers(A), different combinations of constituent phases(B) and ncIn2O3-x(OH)y supported on different substrates(C), proposed activation mechanism for the photocatalytic reaction(D, E)[69]
2.6 氧化物/碳基材料异质结
碳纳米材料(CNM)是现今最有前景的纳米材料之一[70]. 由于sp杂化形成优异的π-π共轭体系,CNM表现出良好的稳定性、 环境安全性和高电导率[71~73], 利用碳材料对光催化剂改性, 已经引起了光催化领域研究学者的极大兴趣[74].
研究人员利用CNM 的独特尺寸、 表面积和结构特性已进行了很多光催化的研究, 在改善CNM 和一些碳基复合催化剂的光催化性能方面取得了较大进展. 如Ahmad等[75]系统总结了碳材料提高ZnO基材料的光催化活性的作用机理(图19): (1) 碳材料的高表面积促进产生更多的反应表面和氧化还原反应位点[76]; (2) 碳材料在ZnO上的锚定实现了电子从ZnO表面到碳材料上的转移, 强化了光生载流子的分离, 并有效抑制其复合[77]; (3) 碳材料与ZnO 异质结构的形成使光学带隙变窄, 拓宽了光吸收范围, 提高了光利用率. 同时, 碳材料的保护减少了ZnO的光刻蚀, 一定程度上提升了催化剂的稳定性.

Fig.19 Advantages of carbonaceous material modified ZnO composite photocatalyst[75]
Zhang 等[78]通过水热路线制备了ZnO/N-rGO 光催化剂. 与纯ZnO 相比, 复合材料具有更高的比表面积, 增强了CO2的吸附能力, 同时异质复合材料具有强的光捕获能力和快速的电荷分离能力, 在加入了CO2和H2O蒸汽为原料的气固反应体系中, 其液态产物CH3OH的生成速率提高4.7倍. Zhu等[79]通过结合2D g-C3N4和Co3O4, 成功地开发了一种典型的二维异质结构, 并将其用于光催化CO2还原反应. 其电荷转移行为和CO2还原位点如图20(A)所示. 引入窄带隙的Co3O4可以拓宽光吸收范围, 使更多的太阳光被响应. 此外, 两种组分之间的紧密接触有效提高了电荷迁移, 并且Co3O4的特定暴露晶面使光生电子通过二者界面迁移到2D g-C3N4, 从而提高光催化反应的表面电荷密度. 因此, 这种2D异质结的构建可以诱导CO2在加入了三乙醇胺的液固体系下高效还原为CO, 其CO 生成量为419 μmol·g-1·h-1, 选择性为89.4%. 该工作充分展示了通过优化合成方法和界面工程来合理设计基于异质结构的光催化剂对提升CO2光还原性能的重要作用和良好前景. Li 等[80]通过简单的浸渍-焙烧工艺, 将WO3纳米颗粒(WO3NPs)附着在g-C3N4纳米片(CN)上形成WO3/g-C3N4(WO/CN)异质结[图20(B)], 可用于三乙醇胺存在下液固相光催化CO2还原. 研究表明, WO/CN异质结的构建有利于光催化剂中光生载流子的迁移和分离过程. 在可见光照射下, 10-WO/CN 的CO 和CH4的生成速率分别提升至单组分催化剂(CN)的8.6 倍和7.5 倍, 同样充分体现出异质结构的优势. 此外, 大量的氧化物和碳基材料复合的异质结构(如NiO/g-C3N4[81], MoO3/g-C3N4[82]和CNT/TiO2[83]等)被制备出来, 均展现出良好的光催化性能.
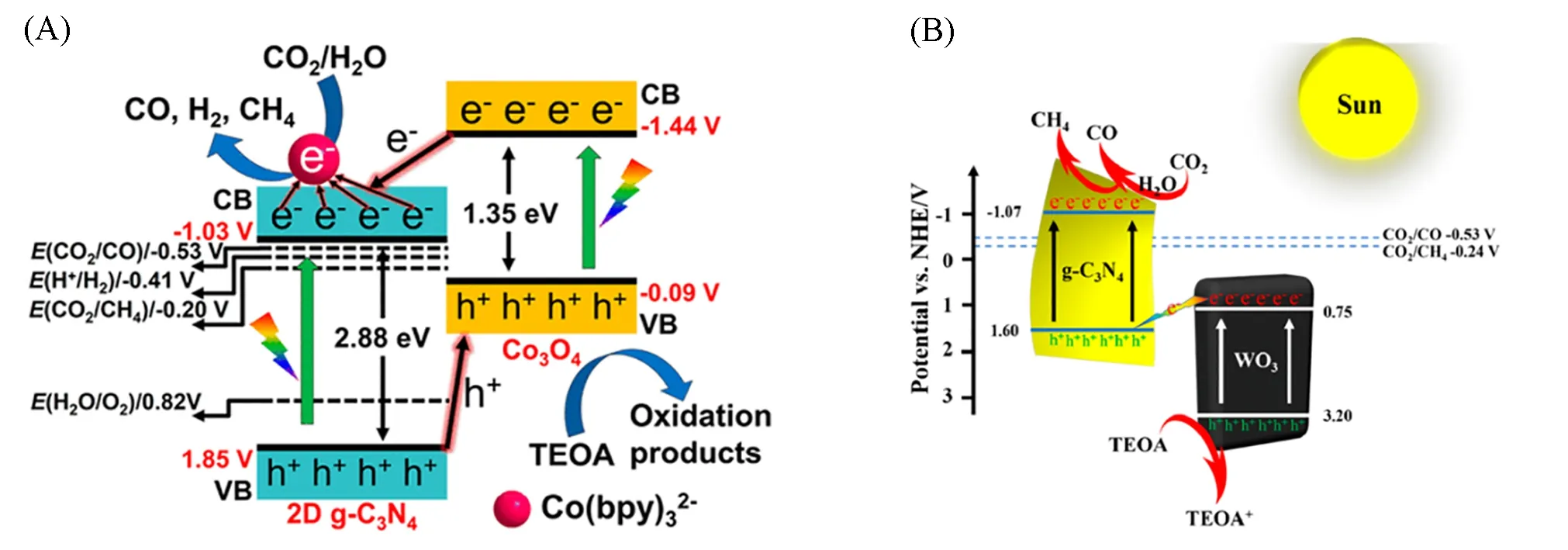
Fig.20 Schematic representation of charge transfer behavior and CO2 photoreduction sites(A)[79] and proposed photocatalytic mechanism of the WO3/CN photocatalyst(B)[80]
3 总结与展望
光催化CO2还原是一种很有前途的方法, 可以利用太阳光将CO2转化为有价值的化学产品和燃料,同时缓解温室效应带来的环境挑战. 设计和制备高效的光催化剂是实现该“人工光合作用”过程的关键. 本文主要综合评述了氧化物异质结构用于光催化CO2还原的最新进展, 系统总结了形成异质结构的类型、 组成等因素, 加深了对提高CO2光催化性能内在机理的认识. 虽然相关研究已经取得了重大进展, 但是仍存在诸多挑战, 值得更加深入的研究.
(1) 目前, 光催化CO2还原仍停留在实验室阶段, 如推向工业应用需解决很多方面的问题. 如: 大多数研究工作使用氙灯作为光源, 只有少数研究采用自然光或模拟太阳光辐照. 而对于较为成熟的氧化物基催化剂体系, 应考虑使用自然光进行半导体材料的光激发. 同时, 应该尽量减少或避免使用牺牲剂(如异丙醇和三乙醇胺), 而尽可能地使用低成本、 无毒和丰富的水作为唯一的还原剂. 另一方面, 目前的大多数研究中使用浓缩的CO2气体, 未来应考虑低浓度CO2的反应环境. 此外, 存在于工业烟气中的氮氧化物和硫氧化物等其它杂质对光催化CO2还原体系的影响是未知的, 须明确和深入理解该因素对催化性能的影响.
(2) 目前, 光催化CO2还原在可见光光照条件下, 不加任何牺牲剂的气固反应体系, 催化产物仍多为CO, 其生成速率已经可以达到毫摩尔每克每小时量级, 选择性超过90%, 并具有数百小时的长期稳定性[84,85]. 但是其催化性能远未达到实际应用的要求, 相对于电催化CO2还原体系和高温热还原体系,仍然效率十分低下. 同时更具附加值的产物(如甲醇、 甲酸盐和C2+产物等), 仍鲜有报道. 具有初步应用规模的光催化CO2还原体系需要将其量子效率达到0.2%以上, 太阳能-光化学能转化效率(STC)超过1%, 同时长期稳定性超过1000 h, 才能具有足够的经济和环境效益. 因此, 设计高效、 稳定且高目标产物选择性的催化材料仍是当务之急.
(3) 深入理解电子-空穴在异质界面间的转移对于在设计助催化剂以促进光催化CO2还原性能方面至关重要. 先进的表征手段(如时间分辨光发射电子显微镜和原位光谱学)可以帮助观察到电子-空穴产生、 分离、 表面迁移和捕捉反应中间体. 此外, 理论计算和实验方法的有效结合可以深入理解和揭示助催化剂在表面反应的真实作用.
(4) 进一步拓展光催化材料体系的选择范围. 目前的研究主要集中在TiO2, ZnO, CuO 和Fe2O3等常规金属氧化物半导体材料, 这些材料的选择主要延续于比较成熟的光解水体系. 在综合考虑成本、稳定性、 安全性等多方面因素的条件下, 需结合光催化CO2还原的自身特点, 有针对性地拓展材料的选择范围. 如在催化剂体系中引入碱性金属元素, 可以提高酸性CO2反应分子的吸附.
