成人先天性肝纤维化3例报告
2023-09-28齐凯凯王铁延何雨倩敖康健
齐凯凯 王铁延 何雨倩 罗 森 敖康健 陈 悦
湖北医药学院附属太和医院感染科 (湖北 十堰,442000)
先天性肝纤维化(CHF)是一种多发生于儿童的常染色体隐性遗传性疾病,是一种罕见的、与胆管板畸形相关的肝内胆管遗传发育障碍疾病[1,2]。本病的发病率极低,有文献报道约为1/10 000至1/20 000[3],而在成年人中更为少见,因其临床表现无特异性,诊断方法主要依靠病理活组织检查,故对于临床诊断CHF尤为困难。本文报道十堰市太和医院感染科收治的3例成人先天性肝纤维化患者。
1 病例资料
病例1:患者,男,54 岁。2020年5月12日,因“腹胀20余天”入住我院,患者20余天前无明显诱因出现腹胀,与进食及活动无关,伴纳差,无其余异常表现。病后曾在当地医院就诊,行腹部CT示:肝硬化,脾大,肝腹水,侧枝循环形成:针对肝硬化来我院就诊,门诊以“肝硬化失代偿期 腹腔积液”收住院。既往史:20余年前曾患梗阻性黄疸;否认其他特殊病史。体检:一般情况尚可,慢性肝病面容,皮肤巩膜轻度黄染,双下肢轻度水肿,余未见明显异常。辅助检查:血常规、肾功能、电解质、胃蛋白酶原等大致正常;肝功能:ALB 33.6 g/L,TBil 71.4 μmol/L,DBil 19.5 μmol/L,IBil 51.9 μmol/L,余未见异常;自身免疫性肝病抗体谱提示:抗可溶性肝抗原/肝胰抗原抗体(SLA/LP) 25.86 RU/mL 升高,余未见明显异常;消化道肿瘤标志物、大便常规含隐血、血清铜蓝蛋白(CER)正常;乙肝核心抗体阳性,甲肝、丙肝、戊肝均阴性均未见异常;胃镜示:食管静脉显露,胃底静脉曲张,慢性浅表性胃炎伴糜烂(Ⅱ级);腹部CT增强示:肝硬化,脾大,侧枝循环开放。肝脏穿刺活组织(图1①)提示肝小叶内肝细胞坏死及变性不明显,汇管区明显扩大,可见大量纤维组织增生,包绕肝实质,形成类似假小叶结构,大量小胆管增生,胆管发育不良,部分囊性扩张,可见少量胆栓,散在少量淋巴细胞浸润,考虑先天性肝纤维化。免疫组化结果:HBcAg(-),HBsAg(-),CK7(汇管区大量增生的小胆管+);Masson及网状纤维染色:汇管区明显扩大,大量纤维组织增生。Hp(-)。结合患者临床表现与肝穿刺组织检查结果,诊断为先天性肝纤维化。
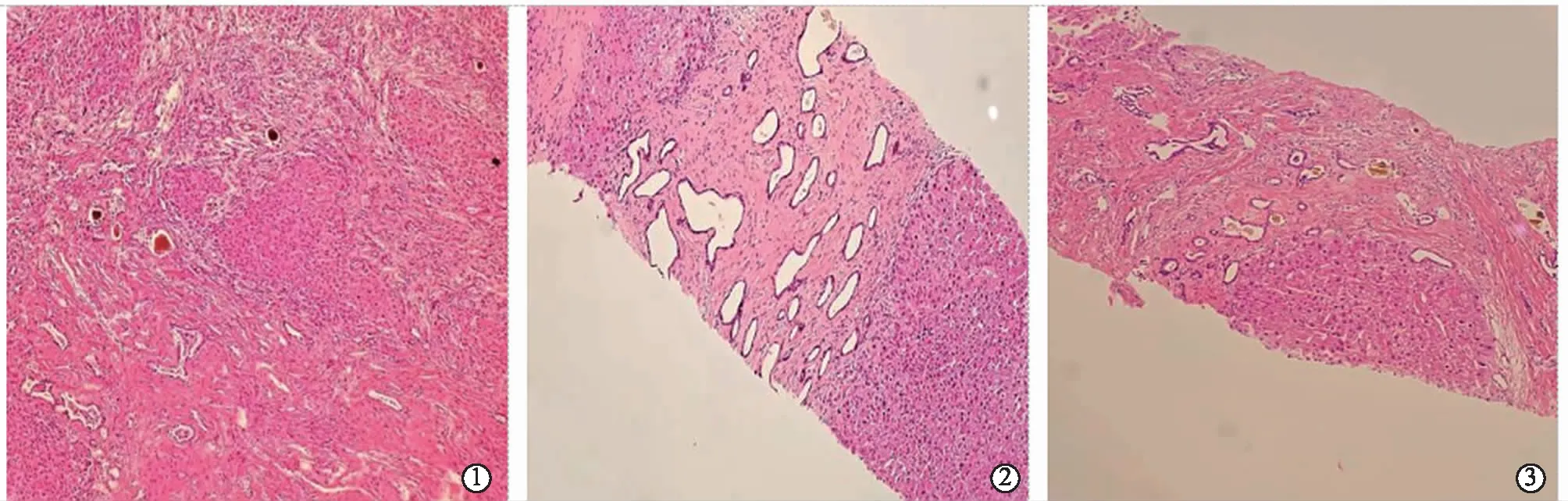
图1 Masson染色,肝脏穿刺活组织检查 (40×)
病例2:患者,女,37岁。2021年11月17日,因“发现肝硬化4年余,TIPS术后3年余”入住我院,患者于2017年在我院住院时发现肝硬化、脾大、门脉高压,完善体液免疫、病毒性肝炎、自身免疫性肝病抗体谱、铜蓝蛋白后,均未见异常,诊断为不明原因肝硬化,给予护肝、降门脉压等对症治疗。于2018-07-02因“肝硬化失代偿期伴胃底食管静脉曲张破裂出血”在局麻下行经皮颈静脉肝内门体静脉分流术,术后予以止血、抗感染、利尿、输血等治疗,患者好转后出院。现患者为求进一步诊治来我院就诊,门诊以“肝硬化、脾大、门静脉高压”收治住院。既往史:胆囊结石7年余,否认其他特殊病史。体格检查:重度贫血貌,肝肋下未及,脾左肋下4 cm,余未见异常。辅助检查:腹部增强CT:①肝脏TIPS术后改变;肝硬化、巨脾,门脉高压症。②食管-胃底静脉曲张出血术后改变。③肝脏、脾脏小病灶(囊肿?);胆囊结石。④腹腔少量积液。血常规:WBC 1.64 G/L,RBC 3.51 T/L,Hb 108 g/L,PLT 28 G/L;肝功能:TBil 23.04 μmol/L,总胆汁酸36.35 μmol/L,余未见异常;凝血功能:PTA 51.4%,PT 16.9 s,INR 1.57,APTT 46.2 s;甲胎蛋白未见明显异常。异常凝血酶原水平为12.91 ng/ml,高尔基体蛋白水平为 145 ng/ml;诊断明确,排除手术禁忌后于2021-11-25在全麻下行3D腹腔镜全脾切除术+肝病损切除术+腹腔镜下腹腔粘连松解术,术后病检提示:(脾脏)慢性纤维淤血性脾肿大;(肝组织)肝脏非肿瘤性病变。肝活检组织(图1②):肝小叶结构紊乱,呈“地图样”,门管区扩大,见广泛纤维化,纤维间隔内散在少量淋巴细胞浸润,见大量增生的小胆管,部分小胆管扩张,小胆管内见胆汁淤积并胆栓形成,网状纤维染色纤维间隔内无网状支架塌陷。病理诊断:先天性肝纤维化。免疫组化结果:CK7(小胆管+),cd38(-),HBcAg(-),HBsAg(-);特殊染色:Masson(+),消化后PAS(-),网状纤维染色(-),亚铁氰化钾(-)。结合患者病史与实验室检查后,考虑先天性肝纤维化。
病例3:患者,男,42岁。2022年9月20日,因“发现肝硬化1年余,肝区隐痛1周”入住我院,1年前体检时发现肝硬化,后反复因“肝硬化”于外院住院,病程中无异常症状,外院检查病毒性肝炎、自身免疫性肝病抗体谱、铜蓝蛋白均无明显异常,肝硬化病因不明确,近1周出现肝区隐痛,为阵发性,放射至右肩部,诉尿黄,无其他不适,病后于济南中医肝病医院就诊,予护肝降酶、退黄治疗,病情无明显好转,为求进一步诊治来我院,门诊以“肝硬化”收入院。既往史:2018年至2019年期间自行口服三七粉;体格检查未见明显异常;肝脏彩超:①肝硬化,门脉高压;②肝囊肿,重度脂肪肝;③胆囊壁毛糙;④脾肿大;⑤少量腹水;肝硬度:34.3 kPa;ANA未见明显异常;肝功能:ALT 40.8 U/L,AST 48.9 U/L,γ-GT 34 U/L,ALP 146.9 U/L,ALB 35.25 g/L,TBil 33.03 μmol/L,25.88 μmol/L,DBil 7.15 μmol/L,凝血功能未见异常,腹部大血管彩超提示三支肝静脉明显变细、血流尚可;下腔静脉未见明显异常;胃底食管静脉扩张;脐静脉开放;腹腔静脉CTV:①门静脉高压。②脾肾分流。2022-09-26行肝穿刺活检,肝穿刺组织病理报告(图1③):肝小叶结构紊乱,门管区扩大,广泛纤维化,粗而宽大的纤维组织分隔肝组织呈假小叶结构,纤维组织内见小胆管增生,管腔大小不一,部分呈小囊样扩张,小胆管内胆汁淤积并胆栓形成,网状纤维染色纤维间隔内无网状支架塌陷,Masson染色示纤维间隔内胶原纤维增生。免疫组化结果:CK7(小胆管+),HBcAg(-),HBsAg(-),MUM1(-);特殊染色:Masson(+),网状纤维染色(-),消化后PAS(+),亚铁氰化钾(+);结合临床表现及肝穿刺检查结果,诊断为先天性肝纤维化。
2 讨论
先天性肝纤维化是一种罕见的肝纤维化多囊性疾病,以常染色体隐性遗传为主[3]。目前大多数学者认为其是多囊肾/多囊肝病变1基因(PKHD1)突变造成的遗传相关性胆管病变[4]。该基因位于人染色体6p21位点上,PKHD1基因的突变会直接影响蛋白fibrocystin/polyductin (FPC)的合成,FPC蛋白表达于再生细胞的初级纤毛,主要分布于肾集合管和袢升粗段以及胆管上皮细胞,缺乏FPC蛋白可导致肾集合管异常囊性扩张和胆管板畸形[5]。FPC蛋白的缺失让未完全成熟的胆管持续进行非特异性炎症坏死向修复的转换过程,从而形成畸形的胆管,此过程中产生大量的胶原纤维沉积于门静脉周围,致使门静脉及其分支受压。伴随着渐进性门脉纤维化形成节段性扩张、胆管畸形所致的胆汁淤积不断进展直至出现相关临床症状。PKHD1基因不仅是目前所知人类常染色体隐性遗传多囊肾病(ARPKD)的主要致病因素[6],同时也是Carolis病[7]的致病基因;CHF、ARPKD与Carolis病均属于纤维多囊性疾病[8],在CHF的发病过程中,常常伴随上诉两种疾病的同时发生,其中最常合并的是ARPKD,由于这三种纤维多囊性疾病不仅在临床表现高度相似而且在病理形态相互重叠,目前多数学者认为他们是同一种疾病的不同表现形式。
CHF是由Kerr等[9]首次命名的一种常染色体隐形遗传疾病。该疾病临床表现无特异性表现,主要表现以门静脉高压、肝纤维化与复发性胆管炎为主,常伴有肾脏囊性疾病[10]。CHF常见于婴幼儿与15岁以下的青少年,成年人发病较为少见,发病无性别差异,可有较为明显的家族史。按照其临床表现的不同,一般分为四种类型:门静脉高压型、胆管炎型、混合型和隐匿型。有文献报道,我国以门静脉高压型最为多见,以上消化道出血、门静脉高压、脾功能亢进、食管胃底静脉曲张为主要临床表现的门静脉高压型占44.4%,以间断性发热、胆汁淤积、腹痛腹胀为主要临床表现的胆管炎型占30.6%,混合型型占8.3%[11]。CHF患者普遍肝实质细胞未受到实质性破坏,肝细胞损伤较小,肝内胆汁淤积情况尚未出现,故在实验室检查中,患者的肝功能情况与其门静脉高压程度并不一致。
CHF患者常以不明原因肝硬化、门静脉高压为首发症状入院,CHF患者无特异性症状,即便影像学与实验室检查对其诊断提供了较为准确的证据,但在临床诊断方面仍较为困难,需要进行病理活组织检查才能进一步确诊CHF。CHF病理表现一般为:肝小叶内肝细胞坏死及变性不明显,肝组织内汇管区明显扩大,可见大量纤维组织弥漫性增生,包绕肝实质,形成类似假小叶结构,但肝中央静脉不受影响,仍处于肝小叶中央区,大量小胆管增生,胆管发育畸形,部分囊性扩张,网状纤维染色纤维间隔内无网状支架塌陷。本文3例患者肝组织内均有大量的致密胶原纤维分布,胆管板畸形明显,有形态各异的未成熟胆管散在分布,这是CHF特异的病理学改变,具有重要的诊断价值[12]。
回顾本文3例患者病史,本组3例患者均为成年人起病且均有肝硬化、脾大、门静脉高压、食管胃底静脉曲张等临床表现,符合CHF中门静脉高压型。针对本文3例患者肝硬化,现分析如下:病例1患者以腹胀为主要症状入院,TBil轻度升高,ALT、AST正常表明肝细胞未见明显炎症坏死,腹部CT有明显门静脉高压表现:腹腔积液,脾大,侧枝循环形成;胃镜提示食管胃底静脉曲张显露,有明显的肝功能情况与其门静脉高压程度不平行的表现,完善病毒性肝炎标志物、自身免疫性肝病抗体谱、免疫球蛋白等相关检查后,提示乙肝核心抗体阳性,进一步完善乙肝DNA测定为阴性,其抗可溶性肝抗原/肝胰抗原抗体(SLA/LP)升高,需要排除自身免疫性肝炎可能,但结合肝脏穿刺活检结果并未发现免疫性肝病相关特征而表现为先天性肝纤维化相关特征,最终诊断考虑为CHF。病例2患者确诊肝硬化4年,并有上消化道出血史,血清胆红素轻度升高,余未见异常,腹部增强CT检查结果提示有明显的门静脉高压表现,结合外院检查结果与肝脏穿刺活检结果,最终考虑诊断为CHF。病例3患者,虽然腹部大血管彩超、腹腔静脉CTV均提示门静脉高压与食管胃底静脉曲张的存在,但临床症状轻微,肝功能轻度异常,排除肝炎病毒感染及其他引起肝硬化因素,最终结合肝脏穿刺活检结果,考虑诊断为CHF。
在CHF的治疗中,尚无特效治疗药物,肝移植是目前唯一的根治性治疗方法[13]。临床上主要处理门静脉高压带来的一系列并发症,缓解疾病的进展是目前的重点,对于CHF患者来说,门脉高压引发的食管胃底静脉曲张破裂出血往往是死亡的首要因素,有研究表明,在早期诊断和干预下,患者预后往往优于其他因素导致的门静脉高压上消化道静脉曲张破裂出血[2]。
综上所述,CHF是由PKHD1基因突变引发的一种常染色体隐形遗传疾病,无特异性临床表现,临床上易漏诊、误诊。其诊断主要靠病理活组织检查,尚无特效治疗药物,肝移植是目前唯一的根治性治疗方案。该病临床上较为罕见,早期诊断与干预尤为重要,临床医生应加强对CHF的重视,以进行早期诊治并改善其预后。
