单核苷酸多态性微阵列技术联合核型分析在颈项透明层增厚胎儿产前诊断中的应用*
2023-09-28童克婷王森林李景然蔡昭方颜宇辉朱健生安徽医科大学妇幼医学中心安徽省妇幼保健院医学遗传中心妇产科合肥30000南方医科大学南方医院肾内科国家慢性肾病临床医学研究中心广东省慢性肾病临床医学研究中心广州510515
童克婷,王森林,李景然,蔡昭方,颜宇辉,朱健生(1.安徽医科大学妇幼医学中心,安徽省妇幼保健院,a.医学遗传中心,b.妇产科,合肥 30000;.南方医科大学南方医院肾内科,国家慢性肾病临床医学研究中心,广东省慢性肾病临床医学研究中心,广州510515)
颈项透明层(nuchal translucency,NT)是指妊娠11~13+6W时在胎儿的颈后部皮肤下的液体生理性积聚,是孕早期产前超声筛查的重要指标[1]。研究发现,NT增厚与胎儿的结构缺陷、染色体异常和遗传疾病相关,会导致不良妊娠结局的发生[2]。染色体核型分析技术是产前诊断中首选的方法,可检出与NT增厚相关的染色体异常等。然而,很多胎儿的遗传性综合征及结构畸形是由于某些染色体微小的不平衡变异导致的,而传统的染色体核型分析技术无法检出。单核苷酸多态性微阵列技术(single nucleotide polymorphism array, SNP array)是一种高分辨率、高通量的全基因组DNA拷贝数变异检测技术,主要用于检测染色体微缺失和微重复,是产前诊断方法中的一线选择[3]。本研究对516例NT增厚胎儿的羊水细胞进行分析,旨在运用SNP array技术联合核型分析探讨胎儿NT增厚与染色体异常相关性,以期为产前遗传咨询及胎儿预后评估提供实验依据。
1 对象和方法
1.1研究对象 选择2020年3月至2023年3月于我院超声科经产前超声检查诊断为胎儿NT增厚的孕妇516例为研究对象,年龄18~45岁,中位年龄29岁。纳入标准:(1)单胎;(2)NT值≥2.5 mm;(3)孕周11~13+6W;(4)行侵入性产前诊断,包括核型分析和SNP array检测。根据NT值分组为:2.5~2.9 mm组183例、3.0~3.9 mm组261例、4.0~4.9 mm组46例和≥5 mm组26例;根据是否合并其他超声异常,分为单纯性NT增厚组360例和合并其他异常指征组156例。本研究经安徽省妇幼保健院医学伦理委员会批准(批准编号:YYLL2022-zd2022-1-2-FA-01),孕妇或其家属均知情同意。
1.2仪器与试剂 LOGIQ E9彩色多普勒超声诊断仪和配套三维超声容积探头(2~5 MHz)购自美国GE公司,原位载玻片培养盒(杭州宝荣公司),羊水培养基(美国Gibco公司),DNA提取试剂(QIAamp DNA Blood Mini Kit,德国Qiagen公司),全基因组扩增试剂盒、Illumina Infinium HD试剂盒(美国Illumina公司),染色体分散仪(美国Thermotron公司),GLS-120全自动染色体核型扫描捕获系统(德国Leica公司),Nanodrop 2000分光光度计(美国Nanodrop公司),杂交炉、洗涤工作站和芯片扫描仪均购自美国Affymetrix公司。
1.3超声检查 由超声医师使用LOGIQ E9彩色多普勒超声诊断仪,配套经腹部三维超声容积探头,探头频率设置为2~5 MHz,检查孕周为11~13+6W,严格按照国际妇产科超声协会(International Society of Ultrasound in Obstetrics and Gynecology,ISUOG)发布的妊娠早期胎儿超声检查指南中的NT值质控标准进行判读,以NT值≥2.5 mm纳入研究。重复测量3次,取其最大值。
1.4侵入性产前诊断 孕妇于妊娠18~26 W时,由具有产前诊断资质的医师在超声引导下行羊膜腔穿刺术,抽取羊水34 mL。其中24 mL羊水经原位培养用于核型分析,10 mL用于SNP array检测。
1.5实验室检测
1.5.1核型分析 用原位培养法对羊水细胞进行培养,收获细胞后,制片染色,通过上机扫描可获得核型信息。用Cytovision核型分析软件计数20个细胞分裂中期染色体核型,并分析5个核型,如有异常核型,增加计数量。核型结果按照国际人类细胞遗传学命名系统(ISCN2020)进行描述。
1.5.2SNP array检测 按照试剂盒说明书提取全基因组DNA,Nanodrop 2000分光光度计检测DNA浓度后,-20 ℃保存。采用Affymetrix CytoScan 750K Array分析平台对羊水细胞DNA进行检测,用Chromosome Analysis Suite(ChAS)2.0分析软件对检测结果进行分析。实验过程按照厂家的标准操作说明进行。所得结果与国际基因组变异数据库(the Database of Genomic Variants,http://dgv.tcag.ca/)对比剔除常见的多态性拷贝数变异(copy number variations,CNVs),其余CNVs通过检索DECIPHER(http://decipher.sanger.ac.uk/)、ISCA(http://dbsearch.clincalgenome.org/search/)、ClinGen(http://www.ncbi.nlm.nih.gov/projects/dbvar/clingen/)、ClinVar(http://www.ncbi.nlm.nih.gov/clinvar/)、OMIM(http://omim.org/)和PubMed(http://pubmed.ncbi.nlm.nih.gov/)等数据库,综合所得数据库报道信息判读CNVs的临床意义。按照美国医学遗传学与基因组学会指南判读拷贝数变异的临床意义[4],将CNVs分为3大类5级:(1)致病性CNVs(pathogenic CNVs, pCNVs);(2)良性CNVs(benign CNVs);(3)临床意义未明CNVs(variants of unknown significance CNVs, VUS-CNVs);(4)可能致病性CNVs(likely pathogenic CNVs, lpCNVs);(5)可能良性CNVs(likely benign CNVs)。
1.6统计学分析 采用SPSS 22.0统计软件进行分析。计数资料采用百分数表示,组间比较采用χ2检验。采用ROC曲线计算曲线下面积,通过敏感性和特异性得到约登指数,从而确定侵入性检查的最佳截断值。以P<0.05为差异有统计学意义。
2 结果
2.1NT增厚胎儿染色体核型分析结果 核型分析技术共检出致病性染色体异常55例(表1),包括47例染色体非整倍体、3例染色体缺失综合征、5例衍生染色体(2号、5号、9号、10号和18号),致病性染色体异常的检出率为10.66%(55/516)。
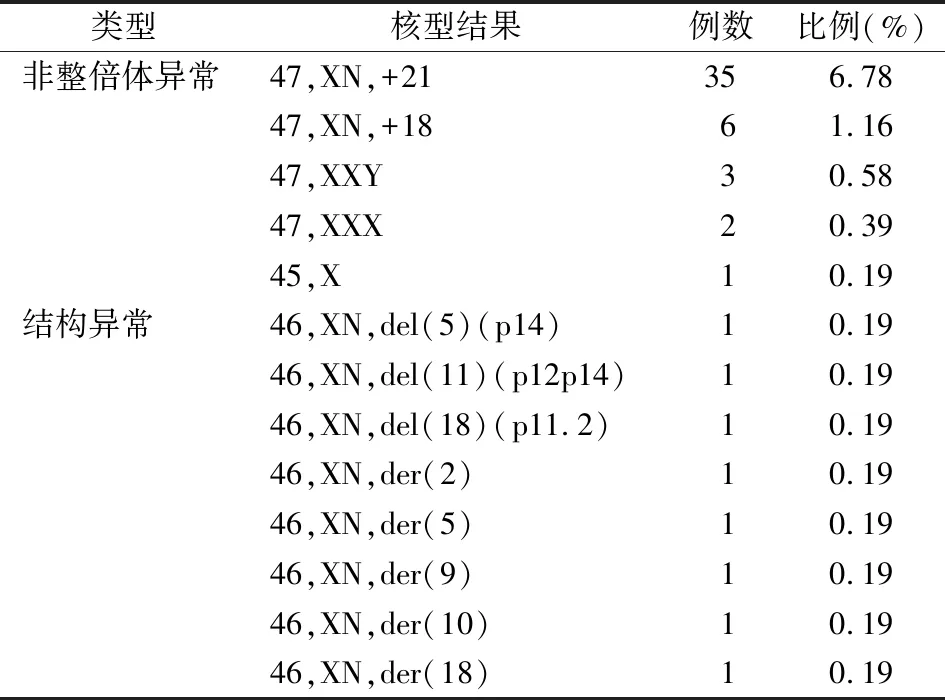
表1 NT增厚胎儿核型分析致病性结果(n=516)
47例染色体非整倍体中,21-三体35例、18-三体6例和性染色体异常6例(克氏综合征3例、特纳综合征1例、超雌综合征2例);3例染色体缺失综合征为猫叫综合征46,XN,del(5)(p14) 1例、18p缺失综合征46,XN,del(18)(p11.2) 1例和WAGR综合征46,XN,del(11)(p12p14)1例;5例衍生染色体为46,XN,der(2) 1例、46,XN,der(5) 1例、46,XN,der(9) 1例、46,XN,der(10) 1例和46,XN,der(18) 1例。核型分析技术还检出7例臂间倒位和8例染色体多态性改变。7例臂间倒位为46,XN,inv(9)(p12q13) 6例、46,XN,inv(18) 1例;8例染色体多态性改变包括46,XN,1qh+ 2例、46,XN,9qh+ 2例、46,XN,13pstk+ 1例、46,XN,15pstk+ 1例、46,XN,15ps+ 1例和46,XN,16qh+ 1例。
2.2NT增厚胎儿SNP array结果 SNP array技术共检出致病性染色体异常70例(表2),包括47例染色体非整倍体(21-三体35例、18-三体6例和性染色体异常6例)和23例pCNVs,检出率为13.57%(70/516)。其中47例染色体非整倍体和3例染色体缺失综合征的SNP array检出结果与核型分析结果基本一致。核型分析检出的5例衍生染色体(2号、5号、9号、10号和18号),经SNP array技术验证,2号衍生染色体是由于2q37.3区域断裂缺失后与18q21.1q23区域重排形成,5号衍生染色体是由于5p15.33p15.31区域断裂缺失后与5p15.31p11区域重排形成,9号衍生染色体是由于9p24.3p23区域断裂缺失后与13q31.1q34区域重排形成,10号衍生染色体是由于10q26.3区域断裂缺失后与4q32.2q35.2区域重排形成,18号衍生染色体是由于18p11.32p11.31区域断裂缺失后与7q34q36.3区域重排形成。另外,SNP array技术还检出48例核型正常的CNVs,包括15例pCNVs(2.91%,15/516)、2例lpCNVs(0.39%,2/516)和31例VUS-CNVs(6.01%,31/516)。核型分析技术额外检出的7例臂间倒位和8例染色体多态性改变,而SNP array未能检出。
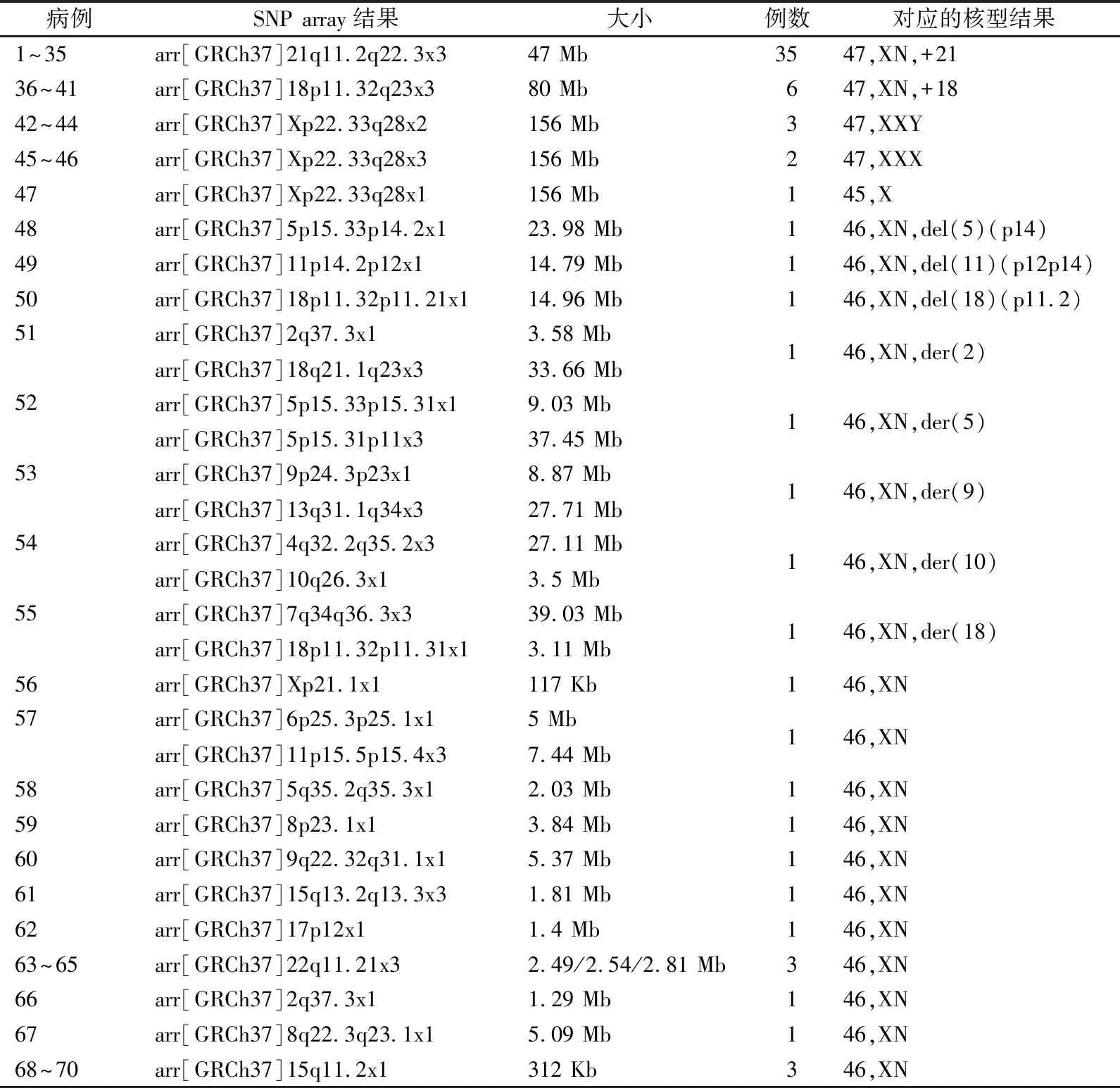
表2 NT增厚胎儿SNP array致病性结果
2.3NT增厚胎儿染色体核型分析与SNP array致病性染色体异常的比较 根据不同的检测技术,将516例NT增厚胎儿分成核型分析组和SNP array组。核型分析技术检出致病性染色体异常55例,检出率为10.66%(55/516);SNP array技术检出致病性染色体异常70例,检出率为13.57%(70/516)。对两组致病性染色体异常的检出率进行统计学分析,差异无统计学意义(χ2=2.048,P=0.152),但与核型分析技术相比较,SNP array技术对致病性染色体异常的检出率要额外多出2.91%(15/516)。
2.4不同NT值分组中胎儿致病性染色体异常的比较 根据NT值,将516例胎儿分成4组:2.5~2.9 mm组、3.0~3.9 mm组、4.0~4.9 mm组和≥5 mm组。2.5~2.9 mm组胎儿183例,致病性染色体异常的检出率为7.10%(13/183);3.0~3.9 mm组胎儿261例,致病性染色体异常的检出率为13.79%(36/261);4.0~4.9 mm组胎儿46例,致病性染色体异常的检出率为23.91%(11/46);≥5 mm组胎儿26例,致病性染色体异常的检出率为38.46%(10/26)。对4组致病性染色体异常的检出率进行统计学分析,差异有统计学意义(χ2=24.472,P=0.000),即随着NT厚度增加,胎儿染色体异常的发生率随之增加。
2.5NT增厚是否合并其他异常指征分组中胎儿致病性染色体异常的比较 根据是否合并其他异常指征(包括高龄、超声异常、血清学异常和不良妊娠史),将516例NT增厚胎儿分为单纯NT增厚组和合并其他异常指征组。单纯性NT增厚组胎儿360例,致病性染色体异常的检出率为8.33%(30/360);NT增厚合并其他异常指征胎儿156例,致病性染色体异常的检出率为25.64%(40/156)。对两组异常染色体的检出率进行比较,差异有统计学意义(χ2=27.805,P=0.000),即NT增厚合并其他异常指征时,其染色体异常的发生率增加(表3)。

表3 NT增厚组是否合并其他异常指征分组中胎儿致病性染色体异常的比较
2.6不同分组中NT增厚胎儿行侵入性检查的截断值 根据是否合并其他异常指征,将516例NT增厚胎儿分为单纯NT增厚组和合并其他异常指征组。采用ROC曲线计算曲线下面积,单纯NT增厚组为0.649(95%置信区间:0.539~0.759,P=0.007),敏感性为0.533,特异性为0.761,约登指数为0.294,最佳截断值为3.46 mm;合并其他异常指征组为0.657(95%置信区间:0.562~0.752,P=0.003),敏感性为0.850,特异性为0.388,约登指数为0.238,最佳截断值为2.95 mm。
3 讨论
本研究运用SNP array技术联合核型分析对516例NT增厚胎儿进行检测,结果显示染色体致病性异常70例,发生率约为13.57%,小于杨培峰等[5]报道的19.3%,这可能是由于地区、样本量大小或入选标准不同而引起的偏差。同时,在各类型的异常结果中以21-三体为最常见,占比6.78%,提示NT增厚对21-三体具有重要的预测价值,这与以往大多数的研究结果相符[6]。黄婷婷等[7]研究表明,随着NT增厚,胎儿染色体异常的发生率逐渐增加。本研究以不同NT值分组为2.5~2.9 mm组、3.0~3.9 mm组、4.0~4.9 mm组和≥5 mm组,胎儿染色体异常的检出率分别为7.10%、13.79%、23.91%和38.46%,与上述研究一致。周林等[8]研究表明,当NT增厚合并其他异常指征时,胎儿染色体异常的发生率明显高于单纯NT增厚。本研究中两组的染色体异常检出率分别为25.64%和8.33%,亦与上述研究相符合。
本研究中核型分析检出致病性异常55例,其中47例染色体非整倍体异常和3例染色体缺失综合征的检出情况与SNP array结果一致。核型分析检出5例衍生染色体,但其分辨率较低,无法识别片段的来源。而SNP array技术可以明确衍生染色体重复或缺失片段的临床意义,是由于相应染色体断裂发生结构重排产生的。核型分析还检出7例染色体臂间倒位与8例染色体多态性改变,而SNP array技术不能识别染色体平衡性变异(如易位和倒位等),同时由于探针的分布情况,其无法检出染色质区、次缢痕、随体以及随体柄等多态性改变。核型分析可以检出染色体数目和形态异常,但无法发现染色体微缺失和微重复[9]。SNP array技术在核型正常的病例中检出2.91% 的pCNVs、0.39%的lpCNVs和6.01%的VUS-CNVs,可以弥补核型分析技术的不足。
目前关于NT增厚截断值的研究尚不统一,国内外大多数以3.0 mm或3.5 mm为NT增厚的截断值[10-11]。但又有相关研究报道,NT值在2.5~2.9 mm范围时,胎儿染色体异常的风险增加[12]。本研究以≥2.5 mm为NT增厚纳入研究,对不同产前指征的NT增厚胎儿进行ROC曲线分析,单纯NT增厚组和合并其他异常指征组行侵入性产前诊断的最佳截断值分别为3.46 mm和2.95 mm。故建议单纯NT增厚胎儿行侵入性检查的截断值为3.5 mm,而当NT增厚合并其他异常指征时,建议其行侵入性产前诊断的截断值为3.0 mm。由于本研究样本量有限,实验结果可能存在一定的偏倚,今后会进一步收集样本进行研究,以期为NT增厚胎儿的产前遗传咨询及预后评估提供更多的实验依据。
