超高效液相色谱-串联质谱法测定动物源性食品中硝基咪唑类药物残留量的方法优化
2023-09-20刘国华何玉榆陈彤谭磊吴雯娟钟名琴
刘国华 何玉榆 陈彤 谭磊 吴雯娟 钟名琴
摘 要:本研究建立了一种超高效液相色谱-串联质谱法测定动物源性食品中硝基咪唑类药物残留量的方法。该方法在硝基咪唑类药物的残留量介于0.300~30.0 ng/mL的范围内时具有良好线性关系,5种硝基咪唑类药物标准曲线的相关系数(r)均大于0.999,方法检出限在4.60×10-2~3.38×10-1 μg/kg范围内,低、中、高不同浓度点加标平均回收率范围分别为74.6%~105.0%、77.4%~106.0%、81.7%~107.0%,相对标准偏差为2.9%~11.7%。试验结果显示,该方法具有快速、简便、准确、灵敏的特点,适用于动物源性食品中硝基咪唑类药物残留量的检测。
关键词:硝基咪唑;动物源性食品;超高效液相色谱-串联质谱法
中圖分类号:S851.33 文献标志码:A 文章编号:1001-0769(2023)04-0096-06
硝基咪唑类药物是一类硝基杂环类抗菌药物,其衍生物易与生物体内的许多大分子物质发生超分子结合[1],在抗厌氧菌[2]、抗寄生虫[3]等方面有广泛的用途。近年来,由于硝基咪唑类药物具有抗原虫和抗菌作用,经常作为饲料添加剂被广泛应用于养殖业,以促进畜禽的生长,提高饲料转化率。然而,在农产品生长期间,以及在饲料和农产品加工、贮藏和运输过程中,均易造成硝基咪唑类药物污染。由于硝基咪唑类药物及其代谢产物具有潜在的致癌、致畸和致突变作用,且代谢物在动物体内能够比原药维持更长的时间[4],如果在动物源性食品中残留、累积,则会造成较大的食品安全风险。因此,硝基咪唑类药物在许多国家被列为违禁药物,我国也对硝基咪唑类药物进行了严格的限制。GB 31650-2019《食品安全国家标准 食品中兽药最大残留限量》[5]明确规定,二甲硝咪唑和甲硝唑可用于动物疾病的治疗,但不得在动物源性食品中检出。GB 31650.1-2022《食品安全国家标准 食品中41种兽药最大残留限量》[6]明确规定,左旋咪唑在禽蛋中的最大限量值为5 μg/kg。其他硝基咪唑类药物在所有食品动物中被禁止使用,且不得检出。
为确保动物源性食品的卫生安全,有必要为残留的硝基咪唑类药物建立一种简单快捷且具有高灵敏度的检测方法。目前,硝基咪唑类药物的测定方法有流动注射化学发光法[7]、酶联免疫吸附法[8]、高效液相色谱 法[9]和液相色谱-串联质谱法等[10-12],虽然目前已有文献报道可用液相色谱-串联质谱法测定动物源性食品中硝基咪唑类药物,但大部分检测方法存在适用对象单一、操作复杂、提取效果差等缺点。传统的国家标准方法,如GB/T 21318-2007 《动物源性食品中硝基咪唑残留量检验方法》[13],利用液相色谱-串联质谱法测定动物源性食品中硝基咪唑类药物,但是采用凝胶色谱净化,该过程繁琐复杂,耗时较长,消耗试剂及材料较多,无法满足当前检测的需要,对操作人员身体健康也可能造成较大危害;农业农村部1025号公告-22-2008[14]只适用于猪的肌肉和肝脏组织,且前处理步骤繁琐,花费时间多,操作不当易造成损失。
本研究对样本前处理方法及仪器条件进行了优化,旨在建立一种简单快捷且灵敏度高的超高效液相色谱-串联质谱法,用于动物源性食品中硝基咪唑类药物残留量的测定,为保障动物源性食品的卫生安全和消费者“舌尖上的安全”提供技术支撑。
1 材料与方法
1.1 仪器与试剂
1.1.1 仪器
试验需要N-EVAP-24型氮吹仪(美国Organomation associates公司)、多管旋涡混合仪(杭州米欧仪器有限公司)、赛多利斯BSA3202S型天平(北京赛多利斯科学仪器有限公司)、SIGMA 3-18K高速冷冻离心机(德国Sigma公司)、超声波清洗器(宁波新芝生物科技股份有限公司)、Aglient 1290超高效液相色谱仪(美国Aglient公司)、配有电喷雾(ESI)离子源的AB Sciex Triple QuadTM
5500 质谱仪(美国AB Sciex公司)等。
1.1.2 试剂与耗材
乙腈(色谱纯)、甲醇(色谱纯)、甲酸(色谱纯)、6cc 200 mg PRIME HLB固相萃取柱(美国Waters公司)等。
1.1.3 标准品
2-甲硝咪唑、羟甲基甲硝咪唑、洛硝哒唑、苯硝咪唑、异丙硝唑:纯度高于95.0%(德国Dr. Ehrenstorfer公司)。
1.2 样本制备
取代表性样本约500 g,使用匀浆机充分捣碎混匀,装入洁净容器中作为试样,将试样密封置于-18 ℃冷冻避光保存。在样本制备时应防止样本交叉污染。
1.3 标准储备液和混合标准工作液的配制
分别准确称取适量2-甲硝咪唑、羟甲基甲硝咪唑、洛硝哒唑、苯硝咪唑、异丙硝唑的标准品,用甲醇配制成浓度为1.00 mg/mL的标准储备液,0~4 ℃冷冻避光保存备用。分别移取适量上述五种硝基咪唑类药物标准储备液,用甲醇逐级稀释成适当浓度的混合标准工作液。
1.4 样本前处理
1.4.1 提取
准确称取样本(2.50±0.02)g于50 mL离心管中,加10 mL乙腈水溶液(含80%乙腈水和0.2%甲酸)后,立即涡旋2 min混匀,用超声波清洗仪超声提取5 min,6 000 r/min高速冷冻离心5 min。
1.4.2 净化
移取上述上清液4.0 mL,直接过PRIME HLB固相萃取柱(6cc 200 mg),保持1 s/滴的速度,收集全部流出液,准确移取2.5 mL流出液至带刻度离心管内。流出液于40 ℃水浴下氮气吹至少于0.3 mL,残留液用甲醇+水+甲酸(体积比为10 ∶ 90∶ 0.09)溶液定容至1.00 mL,涡旋1 min混匀后过
0.22 μm滤膜,供超高效液相色谱-串联质谱仪测定。
1.5 UPLC-MS/MS条件
1.5.1 超高效液相色谱条件
色谱柱:ACQUITYUPLC BEH C18
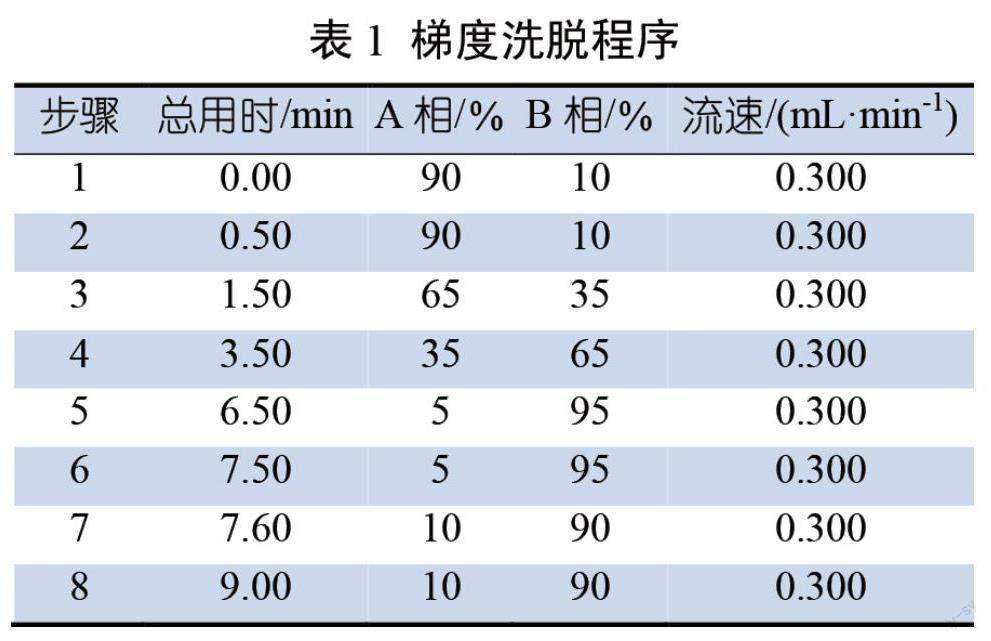
(1.7 μm,3.0 mm×100 mm);流动相A为0.1%甲酸水溶液;流动相B为甲醇(色谱纯);柱温40.0 ℃;进样体积5.00 μL。具体梯度洗脱程序见表1。
1.5.2 质谱条件
电离方式:电喷雾(ESI);电喷雾电压:4 500 V;扫描方式:正离子扫描,多反应监测;离子源温度550 ℃;柱温40 ℃;气帘气压力35.0 psi;碰撞气压力9.0 psi;雾化器压力55.0 psi;辅助加热器压力55.0 psi。
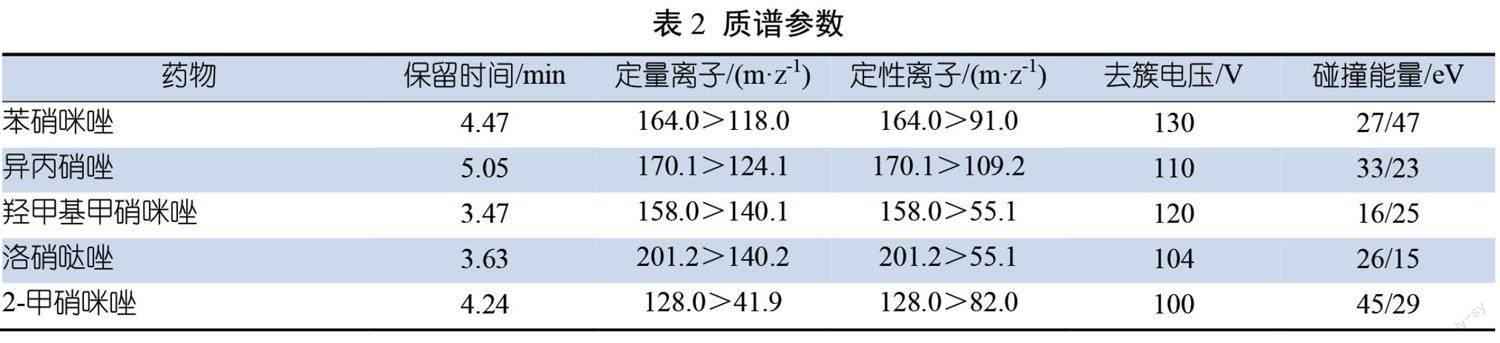
保留时间、定性离子对、定量离子对、去簇电压及碰撞能量见表2。
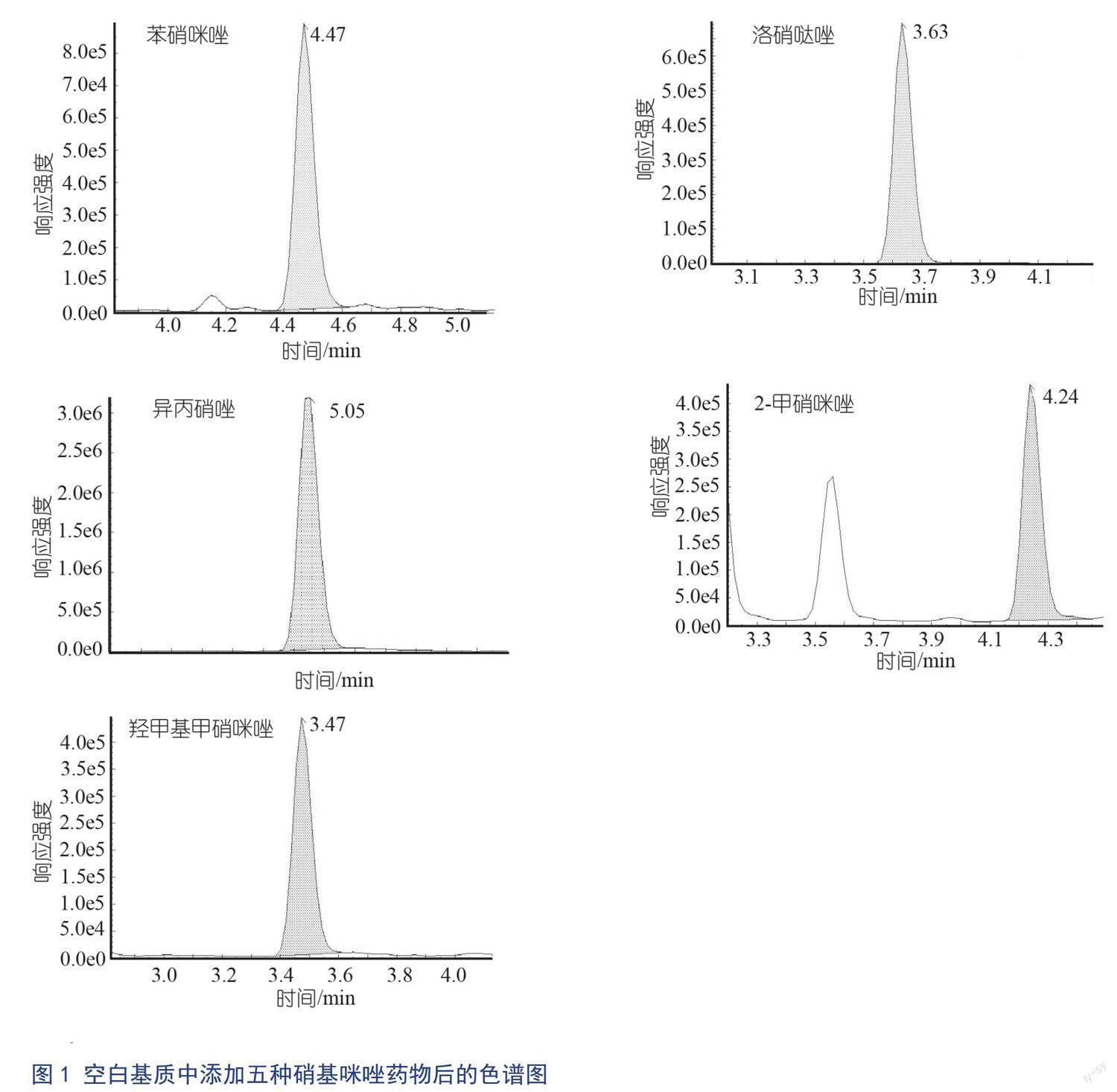
空白基质添加25.0 μg/kg含苯硝咪唑、异丙硝唑、羟甲基甲硝咪唑、洛硝哒唑和2-甲硝咪唑的混合物色谱图见图1。
2 结果与分析
2.1 标准曲线
准确称取5个猪肉空白基质样本,按照1.4所述方法进行前处理,获得空白样本提取液。将空白样本基质配制成不同浓度系列的混合标准工作溶液,羟甲基甲硝咪唑、洛硝哒唑的浓度依次为:0.300 ng/mL、0.600 ng/mL、 1.50 ng/mL、3.00 ng/mL、15.0 ng/mL;2-甲硝咪唑、苯硝咪唑、异丙硝唑的浓度依次为0.600 ng/mL、1.20 ng/mL、3.00 ng/mL、 6.00 ng/mL、30.0 ng/mL。按1.5所述方法设置仪器参数,待仪器稳定后,进行上机测定,绘制标准工作曲线,其中2-甲硝咪唑、羟甲基甲硝咪唑、洛硝哒唑、苯硝咪唑和异丙硝唑的相关系数(r)分别为0.999 2,、0.999 9、0.999 7、0.999 9和0.999 7,可发现基质试验峰面积与相应浓度的线性关系较好,具体见表3。
2.2 方法检出限确定
以空白猪肉样本为基质,做7个平行添加试验,羟甲基甲硝咪唑、洛硝哒唑的阳性添加浓度均为1.00 μg/kg,2-甲硝咪唑、苯硝咪唑、异丙硝唑的阳性添加浓度均为0.50 μg/kg。样本前处理后,进行上机测定。根据各样本检测值计算出平均值( )及标准偏差(SD),计算公式为:
检出限(method detection limit,
MDL)=(k×SD×C)/
式中:k为置信因子,一般取3;C为理论加标水平,单位为μg/kg;测得羟甲基甲硝咪唑、洛硝哒唑、苯硝咪唑、异丙硝唑、2-甲硝咪唑方法检出限分别为4.60×10-2 μg/kg、3.38×10-1 μg/kg、2.24×10-1 μg/kg、1.04×10-1 μg/kg、1.28×10-1 μg/kg(表3)。
2.3 方法回收率和精密度验证
以空白猪肉样本为基质,进行3个不同浓度水平的添加回收实验,每个水平均取6个平行样,2-甲硝咪唑、苯硝咪唑、异丙硝唑阳性添加浓度分别为0.500 μg/kg、 1.00 μg/kg、5.00 μg/kg;羟甲基甲硝咪唑、洛硝哒唑阳性添加浓度分别为1.00 μg/kg、2.00 μg/kg、10.0 μg/kg。按照1.4所述方法进行前处理,处理好的样本供超高效液相色谱-串联质谱测定。猪肉样本中羟甲基甲硝咪唑、洛硝哒唑、苯硝咪唑、异丙硝唑和2-甲硝咪唑的平均回收率和相对标准偏差(RSD)结果详见表4。
由表4可知,本研究所述方法的回收率较好,2-甲硝咪唑、羟甲基甲硝咪唑、洛硝哒唑、苯硝咪唑和异丙硝唑的回收率范围分别为77.3%~107.0%、74.6%~1067.0%、74.6%~106%、82.4%~99.6%和76.6%~98.0%,相对标准偏差范围分别为3.4%~9.5%、2.9%~11.7%、6.0%~7.2%、3.9%~7.1%和4.2%~8.1%,说明所建立的方法重复性好,具有较好的精密度和准确度。
3 结论
本研究建立了一种动物源性食品中硝基咪唑类药物的超高效液相色谱-串联质谱简便测定方法,与传统方法相比,该方法采用酸化乙腈水溶液提取、直通型PRIME HLB固相萃取柱净化的方式,提高了检测效率,检测时间缩减至2 h。该方法灵敏度低、准确度高且分离度好,可适用于动物源性食品中硝基咪唑类药物残留量的定性、定量检测,为硝基咪唑类药物的食品安全风险评估工作提供技术支持。
参考文献
[1] SACKTON K L,DIMOVA N,ZENG X,et al.Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C[J].Nature,2014,514(7524):646-649.
[2] 韩肖亚,陈冬梅,王玉莲,等.几种兽用合成抗菌药基于抗体快速检测技术研究进展[J].中国畜牧兽医,2017,44(6):1869-1876.
[3] 刘得贵,张应龙,杜国辉,等.动物性产品中硝基咪唑类药物残留检测研究进展[J].山东畜牧兽医,2020,41(3):68-71.
[4] 谢文,陈笑梅,丁慧瑛,等.液相色谱串联质谱同位素稀释法对蜂王浆中10种硝基咪唑类药物残留的检测 [J].分析测试学报,2010,29(5):497-501,506.
[5] 中华人民共和国农业农村部,国家卫生健康委员会,国家市场监督管理总局.食品安全国家标准 食品中兽药最大残留限量:GB/T 31650-2019[S].北京:中国标准出版社,2019.
[6] 中华人民共和国农业农村部,国家卫生健康委员会,国家市场监督管理总局.食品安全国家标准 食品中41种兽药最大残留限量:GB/T 31650.1-20-22[S].北京:中国标准出版社,2022.
[7] 谭利.基于分子印迹—流动注射化学发光传感器检测硝基咪唑类物质[D].重庆:重庆师范大学,2018.
[8] 王亚宾.检测硝基咪唑类药物、莱克多巴胺和安定残留的酶联免疫法的建立[D].济南:山东大学,2011.
[9] 于敏,张美娟,张河霞,等.高效液相色谱法测定牛奶中硝基咪唑类残留量[J].食品安全质量检测学报,2018,9(2):447-451.
[10] 张丽媛,周剑,王敏,等.液相色谱-串联质谱法测定鸡肉和鸡蛋中3种硝基咪唑类药物[J].食品安全质量检测学报,2019,10(14):4508-4514.
[11] 魏云计,朱臻怡,冯民,等.高效液相色谱-串联质谱快速测定饲料中硝基咪唑类药物及其代谢物残留[J].分析测试学报,2017,36(3):377-381.
[12] 郑熠,张杉,关旭,等.液相色谱-串联质谱法检测蜂蜜中硝基咪唑类药物残留[J].北京农业,2013(15):147-150.
[13] 国家质量监督检验检疫总局,国家标准化管理委员会.动物源性食品中硝基咪唑残留量检验方法:GB/T 21318-2007[S].北京:中國标准出版社,2007.
[14] 中华人民共和国农业部.动物源食品中4种硝基咪唑残留量检测:中华人民共和国农业部1025号公告-22-2008[S].北京:中国标准出版社,2008.
