金黄色葡萄球菌肠毒素中毒的免疫治疗策略*
2023-08-14窦磊娜于雪芝余文博沈建忠王战辉
李 青 窦磊娜 温 凯 于雪芝 余文博 沈建忠 王战辉
(中国农业大学动物医学院,动物源食品安全检测技术北京市重点实验室,北京 100193)
金黄色葡萄球菌(Staphylococcus aureus)是一种生存能力极强的革兰氏阳性机会致病菌,可产生多种毒力因子,引起广泛感染、中毒性休克综合征(toxic shock syndrome,TSS)和食物中毒。其中,金黄色葡萄球菌肠毒素(staphylococcal enterotoxins, SEs)是金黄色葡萄球菌的主要毒力因子。同时还有一类与SEs结构十分相似但不具有呕吐活性的蛋白质分子。国际葡萄球菌超抗原命名委员会(international nomenclature committee for staphylococcal superantigens,INCSS)将具有呕吐活性的蛋白质分子命名为SEs,缺乏呕吐活性或未被检测是否具有呕吐活性的蛋白质分子命名为类肠毒素(staphylococcal enterotoxins-like,SEls)[1]。
金黄色葡萄球菌的适宜生长温度和pH 值范围较广,因此该菌不仅可以引起浅表皮肤感染和深层组织感染,还可以污染各种各样的食物,包括肉类、蔬菜、水果、糕点和奶制品[2],其分泌的SEs可引起广泛感染和食品污染。SEs 具有高度稳定性,对高温和大多数蛋白水解酶都具有强烈的抗性。金黄色葡萄球菌的菌体细胞在80℃下经30 min 即可被杀灭,而SEs 可耐受100℃高温,即使煮沸30 min 还维持其生物活性和免疫活性,并且SEs能够抵抗胃肠液中胃蛋白酶、胰蛋白酶、木瓜蛋白酶等蛋白酶的水解作用[3-5]。
SEs污染范围广、稳定性高、具有致命的毒性作用,研发出有效的治疗策略十分必要。目前还没有针对SEs治疗策略展开论述的综述。因此本文首先介绍了SEs 的分类,并针对出现频率高、毒性大、 危害最严重的两种SEs—— 肠毒素A(staphylococcal enterotoxin A,SEA)和肠毒素B(staphylococcal enterotoxin B,SEB),总结了其基本结构、超抗原活性及呕吐活性等毒性作用,重点讨论了SEA、SEB 发挥作用的T 细胞受体(T cell receptor,TCR)-SEs-主要组织相容性复合体(major histocompatibility complex,MHC) II 三元复合物的相互作用结合区域以及关键结合位点,并针对SEA、SEB总结了以疫苗为主的主动治疗策略以及以中和性抗体为主的被动治疗策略,以期为SEs有效治疗提供思路。
1 金黄色葡萄球菌肠毒素的分类及结构特点
1959 年,Bergdoll 和Casman 首次发现SE,并将其命名为SEA,至今共发现了23 种SEs 和金黄色葡萄球菌类肠毒素(staphylococcal enterotoxinslike,SEls)[6-10]。这些毒素是由168~261 个氨基酸组成的球状单链蛋白,分子质量大小为19~29 ku。几乎所有的金黄色葡萄球菌都至少编码1 个SE,有些甚至可编码12 个SEs[11]。根据SEs 的核苷酸和氨基酸序列的同源性,SEs和SEls可以分为5组,I 组包括毒性休克综合征毒素1 (toxic shock syndrome toxin-1,TSST-1)、葡萄球菌超抗原样蛋白7 (staphylococcal superantigen-like protein 7,SSL7) 和SElX;II 组包括SEB、SEC、SEG、SElU和SElW;III组包括SEA、SED、SEE、SEH、SElJ、SEN 和SEP;V 组包括SEI、SEK、SEL、SEM、SEQ、SET 和SElV;IV 组来源于链球菌,故本文不予讨论。
尽管SEs之间的一级序列不同,但三维结构十分相似,主要由氨基端和羧基端两个结构域组成[12-13](图1)。氨基端有一个特征性的寡糖/寡核苷酸折叠(oligosaccharide/oligonucleotide fold,O/B fold),形成β 桶状结构,其分子内部高度疏水,但表面覆盖着大量的亲水残基。羧基端含有一个β抓握基序(β-grasp motif),两个结构域之间的α螺旋在分子顶端形成一个与TCR 结合相关的浅腔。以III 组的SEA 和II 组的SEB 为例,具体介绍SEs的二级结构[14-15]。SEA 的氨基端包含两个β 折叠(β-sheet),一个β 折叠由β 折叠股(β-strand)β1、β4a 和β5b 构成,另一个β 折叠由β 折叠股β2、β3、β4b 和β5a 构成,两个β 折叠构成一个β 桶状结构(图1b)。β 桶状结构上被β3 和β4a 之间的α 螺旋(a)SEA(PDB ID:1SXT)、SEB(PDB ID:3SEB)、SEC2(PDB ID:1STE)、SEC3(PDB ID:1JWM)和SEE(PDB ID:5FKA)的序列比对:颜色越深表示氨基酸同源性越大;(b,c)SEA(b)和SEB(c)的三维结构以及活性位点分布,不同的颜色表示不同的二级结构:红色为螺旋,青色为β折叠,绿色为转角,银色为卷曲,黄色为半胱氨酸,虚线为半胱氨酸间的二硫键。(α-helix)α3 覆盖。羧基端包含由β 折叠构成的β抓握基序。两个半胱氨酸残基(Cys96 和Cys106)在β桶状结构底部形成一个二硫键。SEB氨基端包含由β折叠股β1、β2、β3、β4、β5组成的β桶状结构和α2、α3、α5 等3 个α 螺旋(图1c)。两个半胱氨酸残基(Cys93 和Cys113)在β 桶状结构顶部形成一个二硫键。羧基端含有一个由β折叠股β6和β折叠股β12构成的反平行β折叠。SEA和SEB都含有一个由上述二硫键构成的半胱氨酸环,该环被认为与呕吐活性有关[16]。SEA、SEC2和SED的结构中存在Zn2+,其在SEs 与MHC II 类分子相互作用中发挥着必不可少的作用[17-18],但SEB、SEE等与MHC II类分子相互作用时不依赖该Zn2+。
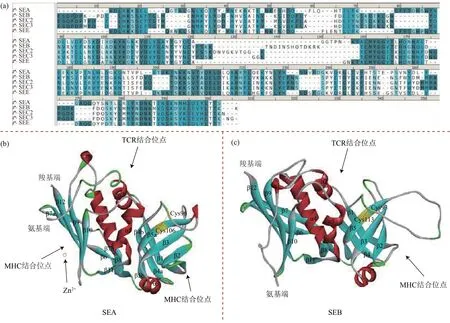
Fig. 1 Primary and tertiary structure of SEs图1 SEs的一级结构和三级结构
2 金黄色葡萄球菌肠毒素的毒性作用
SEs具有多种生物学活性,主要包括超抗原活性、诱导机体呕吐及胃肠炎活性。当SEs通过胃肠道或皮肤创口进入机体,这些生物学活性在SEs产生毒性作用过程中发挥了巨大作用。超抗原与传统抗原刺激T细胞机制不同(图2)。传统抗原经抗原提呈细胞(antigen-presenting cell,APC)内化或处理后形成短的蛋白质水解肽,可与MHC I 或II类分子的多肽结合槽[19]、TCR 的高变区形成特异性的TCR-肽-MHC 复合物[20](图2a)。而超抗原是完整的天然蛋白质,不经APC 的递呈与处理,可直接与MHC II 类分子多肽结合槽以外的位点结合[21](图2b)。并且超抗原绕过与多肽结合的TCR特异性决定区,与特异性的TCR结构域结合,每一种超抗原都对应一种特异性的TCR 结构域[22]。超抗原与MHC II类分子、TCR的这种作用方式导致更多的T 细胞被刺激,释放大量细胞因子[23]。SEs可以激活高达30%的T细胞,导致肿瘤坏死因子-α(tumor necrosis factor α,TNF-α)、白介素(interleukin, IL) -6、 IL-2 和γ 干扰素(interferon γ,IFN-γ)等细胞因子大量释放,最终导致机体产生TSS 甚至败血症[24]。其中SEB 是超抗原活性最强的SEs,雾化SEB可导致动物肺部产生肺水肿和呼吸衰竭[25]。SEB在20世纪60年代被美国列为进攻性生物战剂,在2000 年被美国国家过敏和传染病研究所列为B 类重点病原体[26]。SEB对公共卫生安全产生了巨大威胁。
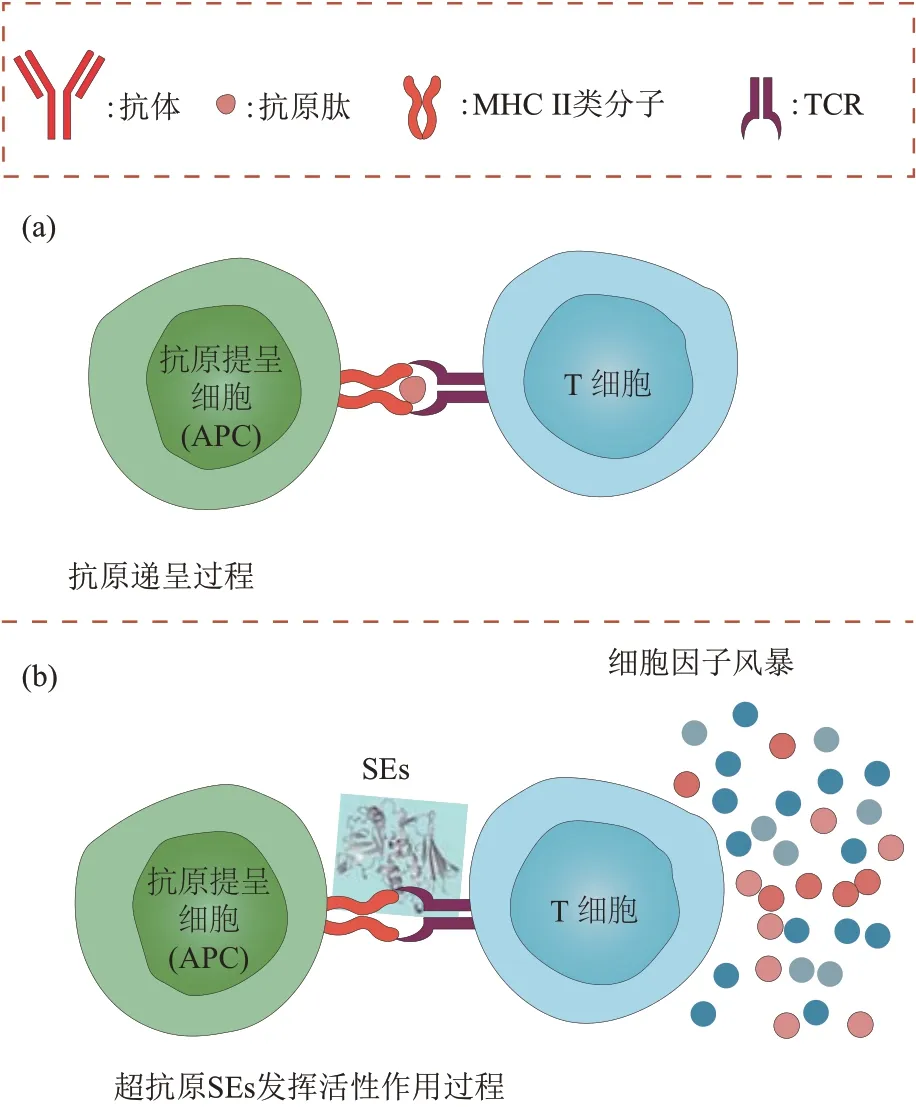
Fig. 2 TCR,MHC class II molecules interact with antigens and superantigens图2 TCR、MHC Ⅱ类分子与抗原、超抗原相互作用
人和动物肠道摄入高纳克或低微克数量级浓度的SEs,会产生发烧、剧烈恶心呕吐、腹痛和腹泻等症状[27-28]。但SEs 导致机体呕吐的机制仍不清楚,有学者认为,SEs导致呕吐主要是引起机体产生5-羟色胺进而刺激迷走神经,但该假说并未得到进一步确证[29]。据报道70%的肠毒素性食物中毒(S. aureusfood poisoning,SFP)都是由SEA 引起的[30]。鉴于SEA 和SEB 的较强毒性,临床主要围绕SEA和SEB的中毒治疗开展研究。
3 TCR-SEs-MHC Ⅱ类分子相互作用位点分析
SEs 主要与MHC II 类分子、TCR 形成TCRSEs-MHC II 类分子三元复合物发挥超抗原活性,因而了解SEs 与MHC II 类分子、TCR 相互作用的关键位点,对于围绕这些关键作用位点开展针对性的研究,达到治疗目的具有重要意义。
所有TCR-SEs-MHC II 类分子三元复合物存在两个相互作用界面,TCR-SEs 相互作用面和SEs-MHC II 类分子相互作用面,而TCR-MHC II 类分子相互作用界面在不同SEs 分子中存在情况不同[31-32]。现有研究多是针对TCR-SEs、SEs-MHC II类分子两个作用面制备疫苗或中和抗体,因此本文围绕SEA、SEB,针对这两个作用面的作用位点展开论述。
3.1 TCR-SEs相互作用位点分析
不同的SEs识别不同的TCR亚型,与SEs相互作用的TCR 结构域具有特异性[22]。SEA 与人TCR亚型Vβ5.2、Vβ5.3、Vβ7.2、Vβ9、Vβ16、Vβ18 和Vβ22 链均存在相互作用[22]。SEB 可与人TCR 亚型Vβ3、Vβ12、Vβ14、Vβ15、Vβ17 和Vβ20 链结合[15]。人TCR Vβ利用互补决定区(complementary determining region, CDR) 2、 骨架区(frame region,FR) 3、高变区(hypervariable region,HV)4和CDR1与SEA羧基端与氨基端之间的浅槽相互作用,分别占SEA-TCR 复合物中的TCR 埋藏面积的37%、20%、23%和10%。SEA 有4 个区域与TCR产生相互作用(图3a),包括α2螺旋(残基Gly20、Thr21、Gly24、Asn25、Lys27 和Tyr32、Asn33、Glu34),β2-β3 和β4-β5a 环组成的疏水区(残基Ser62、Trp63、Tyr64、Tyr91、Tyr92、Gly93和Tyr94),α4-β9 环(残基Val174 和Phe175),以及α5 螺旋的氨基端一侧(残基Tyr205 和Ser206)[33]。人TCR与SEB氨基端β桶状结构和α2螺旋之间的浅槽结合。SEB 中的残基Asn23、Val26、Asn31、Val33、Asn60、Tyr61、Tyr90 和Tyr91残基在毒素分子与人TCR相互作用过程中均发挥了重要作用(图3b)[32,34]。小鼠TCR Vβ8.2结构域上FR2 的残基His47,CDR2 的残基Tyr50、Ala52、Gly53、Ser54和Thr55,FR3的残基Glu56、Lys57、Tyr65、Lys66 和Ala67,HV4 的Pro70 和Ser71,都是与SEB相互作用的重要位点[35]。
3.2 SEs-MHC II类分子相互作用位点分析
MHC II 类分子与不同的SEs 分子结合时的位点不同,但存在部分重叠[36]。一些SEs 结合MHC II类分子的α1区域,一些SEs结合MHC II类分子的β1 区域,一些SE 与MHC II 类分子的α1 区域和β1区域同时结合。
SEA 既可与MHC 的α 或β 同时结合,但一个SEA分子不能与同一个MHC II类分子α、β同时结合,可以与两个MHC II 类分子的α 或β 同时结合[28]。MHC II 类分子β 链的His81 对SEA 的结合很重要[37]。Hudson 等[38]发现了在与MHC II 类分子的相互作用过程中,SEA的两个作用位点。第一个高亲和力位点是由Zn2+介导的His187、His225、Asp227 三个关键氨基酸与MHC II 类分子β 链中的His81 相互作用。第二个低亲和力位点的相互作用与SEB 类似,为Phe47 与MHC II 类分子α 链相互作用。第一个高亲和力位点的相互作用可增强第二个低亲和力位点结合。这两个结合位点是SEA 发挥最大活性所必须的,两个位点的存在使得SEA能够跨越APC 上的MHC-II 分子,增加超抗原活性[39]。
SEB 与MHC II 类分子α 链相互作用[28]。SEB与MHC II 类分子的结合面由SEB 的两个保守结构组成,包括由β 桶状结构的3 个β 折叠股1(残基Val33-Lys39)、2(残基Asp48-Ser52)和3(残基Asn63-Phe68)衍生的极性结合袋和疏水性β转角。另外,以亮氨酸残基为中心的疏水环在大多数SEs中是保守的(SEA-Leu48,SEB-Leu45),是与MHC II 类分子相互作用的关键残基之一[36,40]。SEB 通过残基Phe44 和Leu45 结合MHC II 类分子α1 区域的保守疏水结合袋[41]。当突变了SEB 分子中残基Phe44和Leu45时,SEB与MHC II类分子的结合亲和力被破坏[34]。SEB 的结合袋残基Glu67、Tyr89 和Tyr115,结合MHC II 类分子α 亚基的Lys39。
SEA、SEB 利用类似的蛋白质结合基序与MHC II 类分子发生相互作用,结合基序周围的结构对MHC II 类分子与SEs 的结合亲和力也至关重要[42]。SEs 的极性结合袋与二硫键合环等保证了SEs-MHC II类分子复合物结合的稳定性与特异性。SEA中有一个短二硫键合环,SEB中有一个长的同源长环。SEB长环中的残基Tyr94与MHC II类分子α 亚基的Leu60 和Ala61 形成疏水相互作用,SEA中的Ala97具有类似于SEB的Tyr94的作用。
4 金黄色葡萄球菌肠毒素中毒的免疫治疗策略
目前尚没有针对SEs引起的致死性休克的有效疗法。免疫疗法是预防和治疗金黄色葡萄球菌相关疾病的一种非常有前景的治疗策略[43]。针对SEs开展的治疗性研究也大多数是围绕抗体的主动免疫疗法和被动免疫疗法,主动诱导或被动给予机体针对SEs的中和性抗体(图4a,b)。免疫疗法产生的中和性抗体可以特异性地结合SEs,中和SEs 的毒性作用,减少促炎细胞因子的产生,从而产生保护作用。主动免疫疗法通常选择SEs发挥超抗原活性的某个表位,消除SEs的超抗原活性,用其制备疫苗,诱导机体产生保护作用的抗体(表1)。被动免疫疗法通常是给予机体中和性抗体,这些针对SEs 的中和性抗体主要分为两类,一类是可识别MHC II 类分子结合表位的抗体,一类是可识别TCR 表位的抗体(表2)。这两类抗体都通过破坏TCR-SEs-MHC II 类分子三元复合物的形成从而阻断SEs的超抗原作用以达到治疗目的。

Table 1 Research on active therapy for SEs表1 主动疗法治疗SEs的研究

Table 2 Research on passive therapy for SEs表2 被动疗法治疗SEs的研究
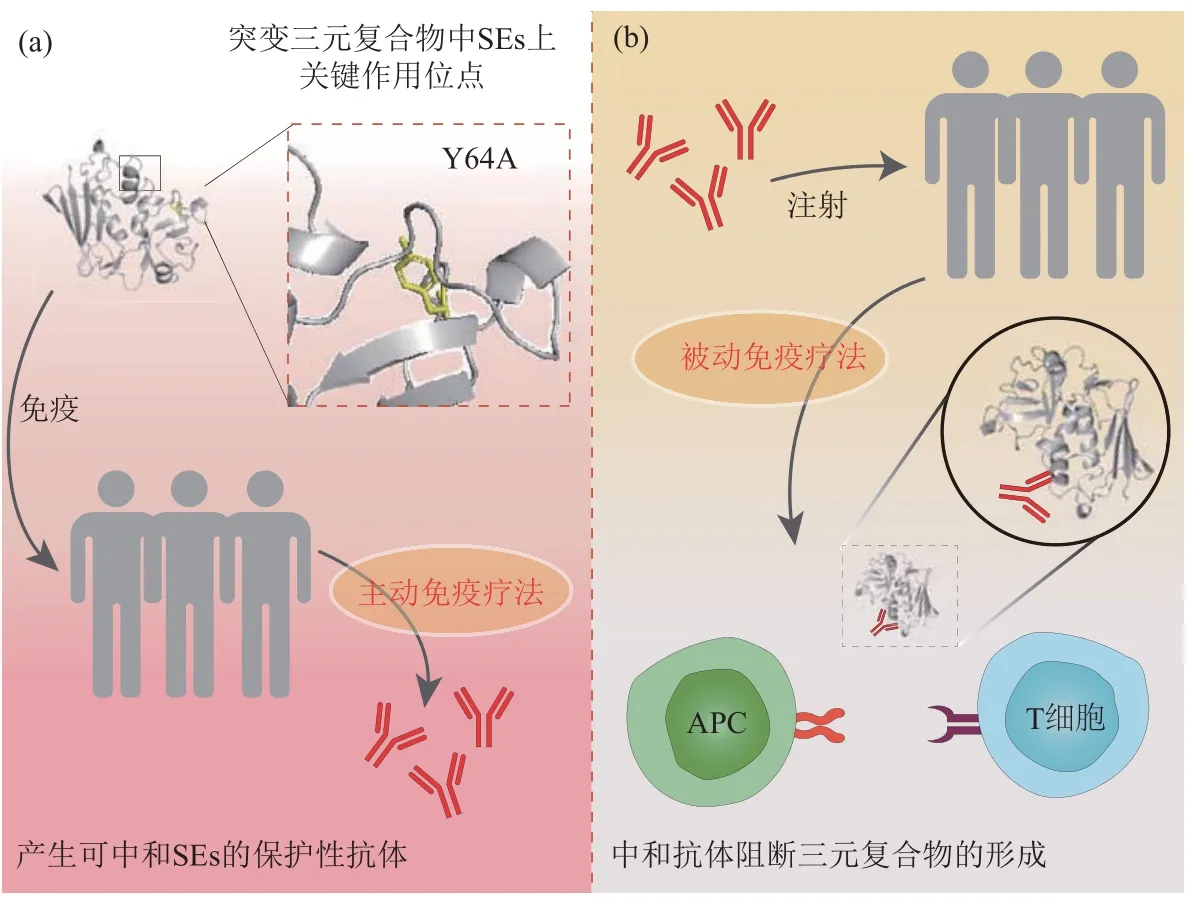
Fig. 4 Active and passive immunotherapy for SEs图4 主动免疫疗法和被动免疫疗法治疗SEs
4.1 主动免疫疗法在预防金黄色葡萄球菌肠毒素中毒中的应用
4.1.1减毒疫苗
1996 年,Bavari 等[44]通过定点突变分别将SEA 上的64 位和92 位酪氨酸突变为丙氨酸(SEAY92A,SEAY64A),从而破坏TCR-SEA-MHC II类分子三元复合物的形成,将突变后的SEA 作为疫苗免疫小鼠。SEAY92A 降低了SEA 与HLA(human leukocyte antigen)-DR1 结合能力;SEAY64A降低了SEA与TCR的相互作用;体外实验表明,SEAY92A和SEAY64A不能诱导T细胞增殖。高剂量SEAY92A 或SEAY64A 免疫的小鼠可完全抵抗WT SEA毒素攻击,所有接种疫苗的小鼠血清中均检测到较高水平的SEA 抗体。1998 年,该团队发现,通过单一定点突变SEB 极性结合袋(Y89A、Y115A、E67Q) 或疏水结合环(Q43P、F44P、L45R)上的关键氨基酸残基,构建的重组SEB 可以破坏SEB与HLA-DR1的结合[45]。这些突变体对T细胞无刺激性,不引起细胞因子生成,所有免疫小鼠均产生了持续时间较长的免疫应答。该疫苗在灵长类动物身上同样有很好的保护作用,接种重组SEB 的小鼠可以承受WT SEB 10~30 倍的LD50攻击,接种重组SEB 的非人灵长类动物恒河猴可以承受30 倍的LD50攻击。此外,该疫苗具有一定的交叉保护作用,可以引起较高水平的SEC1抗体产生,以及一定程度的SEA、TSST-1 抗体产生。随后,Boles 等[46]将SEB 的MHC II 类分子结合位点的3个氨基酸突变(SEBL45R/Y89A/Y94A),制备了一种重组SEB 减毒疫苗STEBVax。该减毒疫苗在小鼠模型中产生了良好的保护作用。将该减毒疫苗免疫恒河猴后,使用致死浓度的雾化SEB 攻击恒河猴,该减毒疫苗对恒河猴产生了剂量依赖性保护作用。目前,SEB疫苗STEBVax已经完成了临床I期试验,所有志愿者表现出良好的剂量耐受性[47]。2017年,Choi等[48]基于以往研究基础和计算机模拟技术设计了4 个重组SEB 减毒疫苗:S2(SEBF44A/E67A)、 S9 (SEBN23A/Y90A)、 S19(SEBN23A/Y90A/R110A/F177A) 和S26 (SEBF44A/E67A/Y90A/R110A/F177A)。除S2外,其他3个重组SEB 减毒疫苗均不表现T 细胞刺激活性。S9 和S19 在小鼠模型中具有100%的保护作用,而S26仅产生20%的保护作用。其原因可能是S26的氨基酸取代改变了减毒疫苗发挥诱导中和抗体产生的原始结构,影响免疫效果。
4.1.2亚单位疫苗
2002 年,LeClaire 等[49]制备了SEB 前99 个氨基酸残基的重组SEB 氨基端片段(1~99)和羧基端(66~243)氨基酸残基片段。将这两个蛋白质片段免疫小鼠后发现,两个重组片段具有很高的免疫原性,但产生的抗体没有中和作用,无法保护小鼠免受SEB 的致死性攻击,说明SEB 重组片段不是设计有效疫苗的理想选择。2012 年,Shylaja等[50]构建了一个包含SEB 的融合蛋白(SEBTSST-1),该融合蛋白可诱导机体产生针对SEB的抗体。但该研究并未进一步探究该融合蛋白的疫苗保护作用。随后,Reddy 等[51]评估了一个包含SEA56~177 的融合蛋白(SEA-TSST-1)在小鼠中的保护作用。将该融合蛋白免疫小鼠后15 d 或120 d 给予小鼠4 倍LD100的毒素,小鼠存活率分别为80%和50%,空白对照存活率为0%。可见该疫苗不仅可以诱导机体产生针对SEA 的中和抗体,还可以使机体产生针对SEA 的免疫记忆。Kota等[52]在Shylaja等和Reddy等的研究基础上,设计了一个包含SEA、SEB 和α 溶血素的嵌合蛋白r-HAB,该融合蛋白可以在小鼠体内诱导强抗体反应,在致死剂量的毒素攻击下对小鼠提供了83%的保护作用。用抗r-HAB 抗体被动给予机体,在致死剂量毒素攻击下可以对小鼠提供大约50%的保护率。这些疫苗设计均保留了SEs发挥毒性作用的作用位点,在使用的过程中存在一定的生物安全隐患。Venkatasubramaniam 等[53]设计了一种由TSST-1、SEB 和SEA 突变体构建的毒素融合蛋白——TBA225。首先改造SEA、SEB 和TSST-1 的MHC II 类分子结合面,分别产生3 个突变毒素分子:SEAL48R/D70R/Y92A、SEBL45R/Y89A/Y94A 和TSST-1L30R/D27A/I46A。此前有报道,SEA高亲和力MHC结合位点H225的突变可降低毒素激活T细胞的能力[38]。因此将H225A 作为额外的安全突变引入到SEA 三重突变体中,产生了SEAL48R/D70R/Y92A/H225A。SEA 突变体编码基因与SEB、TSST-1 突变体编码基因融合构建了TBA225。该疫苗在小鼠体内可同时产生针对SEA、SEB和TSST-1的中和抗体。在致死性毒素联合LPS 攻击实验中,TBA225免疫对SEB和TSST-1的保护率为100%,对SEA的保护率为90%。利用TBA225制备的兔多抗对TSST-1、SEA、SEB、SEC-1、SEH、SEK、SEE、SED都具有一定的中和作用。此外,TBA225诱导的抗体可中和包括USA300在内的多个临床相关金黄色葡萄球菌培养上清液对人类外周血单个核细胞(peripheral blood mononuclear cell,PBMC) 的毒性作用。
B细胞表位疫苗不包含毒素蛋白完整结构,生物安全性高,且易于生产。在Boles 等[46]的研究基础上,Zhao 等[54]利用酶联免疫吸附试验(ELISA)又进一步确定了6个可用于制备B细胞表位疫苗的免疫优势线性表位:SEB31~48、SEB97~114、SEB133~150、SEB193~210、SEB205~222 和SEB247~261。将6 个线性表位分别偶联偶联钥孔血蓝蛋白(keyhole limpet hemocyanin,KLH)后免疫小鼠,均在小鼠体内诱导出SEB 特异性抗体,并可对抗甲氧西林金黄色葡萄球菌(methicillin resistantStaphylococcus aureus,MRSA)感染产生部分保护作用,其中SEB193-210-KLH联合弗氏佐剂与其他表位/佐剂组合相比表现出最好的保护作用(70%)。6个线性表位串联融合蛋白免疫后的小鼠在MRSA攻击后,表现出比STEBvax[46](85%或90%)更好的保护效果(90%或100%)。
4.1.3其他疫苗
SEB主要通过黏膜进入宿主,因此与传统接种途径疫苗相比,黏膜接种疫苗以及黏膜抗体IgA在SEs 中毒的免疫治疗中十分重要。Inskeep 等[55]在Boles 等[46]的研究基础上,将STEBVax 疫苗口服给予仔猪模型后,仔猪血清IgG和粪便IgA均可识别SEB。这一现象表明黏膜疫苗不仅能诱导机体黏膜免疫,还能诱导外周IgG 产生。随后,Xiong等[56]以枯草芽孢杆菌为载体,构建了表达STEBVax 的疫苗。与空白对照相比,口服表达STEBVax 的枯草芽孢杆菌孢子的小鼠粪便中出现SEB 特异性IgA,血清中出现SEB 特异性IgG1 和IgG2a。与空白对照相比,口服接种该疫苗的小鼠模型存活率提高33.3%。
溶解微针(microneedles,MNs)是微米级结构体,其针头搭载的靶标分子及构成针头的全部成分均由对人体无害的生物分解性物质组成,通过完全溶解在使用部位内的方式将靶标分子输送到体内任何部位。皮肤中存在大量可用于抗原提呈的朗格汉斯细胞和组织树突细胞,因此Liu 等[57]评估了负载有STEBvax,由硫酸软骨素和海藻糖构成的MNs 免疫效果。该疫苗不仅延长了体内抗原保留时间,还诱导出高水平的SEB 特异性抗体反应,在致死剂量SEB 攻击时可以提供80%的保护率。这一方法为SEs的疫苗制备提供了新的研究思路。
4.2 被动免疫疗法在治疗金黄色葡萄球菌肠毒素中毒中的应用
4.2.1单克隆抗体
近些年来一直有针对SEs 中和抗体的研究报道。2010年,Tilahun等[58]制备了一对识别不同表位的鼠源SEB 中和抗体63.1.1 和82M.1.2,其与SEA、TSST-1 无交叉反应,这对抗体以协同作用抑制SEB 诱导的T 细胞增殖。2014 年,Xia 等[18]制备了亚纳摩尔亲和力的抗SEB 鼠源中和抗体3E2,3E2具有阻断SEB和MHC II类分子相互作用的功能,且3E2与SEA、SEC没有交叉反应。突变分析表明,SEB上的残基Y46和K71是3E2产生中和作用的关键结合残基,其中Y46是3E2区分SEB和SEA所必需的。Drozdowski等[59]利用电融合方法制备出分泌高亲和力SEB MAb 的人源杂交瘤细胞,产生的HuMAb-154 可以抑制SEB 诱导的人原代淋巴细胞促炎细胞因子INF-γ 和INF-α 的分泌。预防给予抗体HuMAb-154 的小鼠可以抵抗高达100 μg 的SEB 攻击,在SEB 攻击后给予该抗体同样也可以提高动物的存活率。
与甲氧西林敏感金黄色葡萄球菌(methicillinsusceptibleStaphylococcus aureus,MSSA) 菌株M11118 相比,MRSA 菌株SEB 编码基因第703 位点都发现一个额外的碱基,从而导致235、236 和238 位的3 个氨基酸发生变化(Y235T、N236T 和Q238K),影响中和性抗体结合[60]。Varshney等[60]将来源于MSSA的全长SEB免疫BALB/c小鼠,获得4 株特异性单克隆抗体(MAbs) ——20B1、14G8、4C7 和6D3。这4 株MAbs 不与SEB 羧基端缺失11 个氨基酸的SEB 结合,表明SEB 羧基端存在介导SEB 与这4 株MAb 特异性结合的B 细胞表位。其中,20B1、14G8 和6D3 与SEB 有纳摩尔的结合亲和力,可以抑制SEB诱导T细胞增殖以及人源T 细胞体外分泌IL-2 和IFN-γ。在BALB/c 小鼠和HLA-DR3小鼠模型(缺失小鼠内源性MHC II类分子,表达HLA-DR3分子以及人源CD4分子)中研究MAb对SEB诱导的致死性休克(SEB-induced lethal shock,SEBILS)的保护作用,发现高剂量MAb 20B1 对两种小鼠模型的SEBILS 均有高保护率,MAb 14G8 对BALB/c 小鼠无保护作用,MAb 6D3 对BALB/c 小鼠仅有部分保护作用,高剂量MAb 14G8 和Mab 6D3 对HLA-DR3 小鼠无保护作用。而同时给予一种保护性和一种非保护性MAb(20B1+14G8 或20B1+6D3)或同时给予两种非保护性MAb(14G8+6D3)对HLA-DR3 小鼠也有保护作用。该团队利用核磁共振和结晶学来研究SEB和MAb 20B1、14G8、6D3之间的相互作用,以确定MAb增强保护效力的机制[61]。结果表明,MAb 20B1和TCR竞争性结合SEB,20B1与SEB的结合亲和力是TCR与SEB结合亲和力的1 000倍,因此阻断了TCR-SEB-MHC II 类分子三元复合物的形成。MAb 14G8和6D3与SEB的结合表位距离TCR和MHC II 类分子表位较远,因此保护作用低。高剂量的MAb 20B1 还可保护MRSA 来源的SEB 中毒。在MRSA 感染的脓毒血症模型中,预先使用MAb 20B1 静脉注射小鼠,MAb 20B1 可与感染组织中的SEB 结合,降低促炎细胞因子水平、淋巴细胞增殖和中性粒细胞募集,同时降低皮肤浅层和深层组织中的细菌负荷,提高动物存活率[62]。
4.2.2基因工程抗体
Tilahun 团队[58]将鼠MAb 的轻链可变区(light chain variable region,VL) 和重链可变区(heavy chain variable region,VH)的编码基因分别移植到编码人Igκ和IgG1恒定区的基因上,制备出可适用于人类的人-鼠嵌合抗体Ch 63 和Ch 82 M。在人PBMC 或HLA-DR3 小鼠脾细胞中进行测试发现,人鼠嵌合抗体Ch 63 和Ch 82 M 以协同作用中和SEB。人鼠嵌合抗体Ch 63 和Ch 82 M可以减弱由产SEB的金黄色葡萄球菌引起的全身炎症反应,在金黄色葡萄球菌肺炎模型中可显著提高小鼠存活率[63]。人鼠嵌合抗体Ch 82 M与具有免疫调节活性作用的洛伐他汀联用时,对小鼠致死性TSS 也产生协同保护作用[64]。洛伐他汀或人鼠嵌合抗体Ch 82 M 单独作用于HLA-DR3 小鼠时,对致死性TSS起到了部分保护作用(分别为50%、66%),而洛伐他汀与Ch 82 M联用时,保护率为100%。临床安全性他汀药物和低免疫原性嵌合抗体的联合使用为人体SEs 中毒治疗提供新思路。
Varshney 等[65]将MAb 20B1 的轻重链CDR 区分别嫁接到人源胚系骨架IGKV1-39 和IGHV3-7,制备了两个人源化SEB抗体Hu-1.6/1.1和Hu-1.4/1.1。这两个人源化抗体对SEBILS 的治疗水平与MAb 20B1 相当,经Hu-1.4/1.1 或Hu-1.6/1.1 处理后的小鼠存活率为80%,而未处理的小鼠存活率为10%。用小鼠金黄色葡萄球菌败血症模型进一步探讨Hu-1.6/1.1 和Hu-1.4/1.1 的保护作用,发现与MAb 20B1以及万古霉素相比,Hu-1.6/1.1可显著提高产SEB MRSA 菌株引起的致命脓毒症小鼠存活率。Hu-1.6/1.1 治疗组小鼠的存活率为90%,而同型对照单抗治疗的小鼠存活率为10%,万古霉素治疗的小鼠存活率为40%。此外,Hu-1.6/1.1 与万古霉素联合治疗进一步提高了动物存活率并改变了细胞因子反应,Hu-1.6/1.1 或万古霉素单独治疗小鼠的存活率为40%,Hu-1.6/1.1 和万古霉素联用治疗小鼠的存活率为90%,这种联合疗效的提高从IFN-γ、IL-10 等细胞因子水平上也得到了体现。在深部组织感染模型中,Hu-1.4/1.1 与SEB 结合后可降低促炎细胞因子水平,缓解组织脓肿。
噬菌体展示技术是筛选SEs中和抗体的重要手段。2010 年,Larkin 等[66]利用噬菌体展示技术,制备出针对STEBVax 的人源抗原结合片段(antigen-binding fragment,Fab)以及其全长IgG,这些人源MAb 可高亲和力、特异性地中和毒素,保护小鼠免受SEBILS。当效力较弱的Fab 转化为全长IgG时,效价可以得到明显提高。Larkin等[66]认为可能是Fc 区域在抗体中和作用中发挥了较大作用。但MacIntyre 等[67]将Fc 区域进行突变后证明,SEB的中和不需要Fc受体结合发挥中和作用。全长IgG中Fc的存在可能通过增加Fab的构象稳定性,进而提高抗体效价。2012年,Karauzum等[68]利用基于噬菌体展示技术筛选出一组高亲和力的SEB 人源Fab。将Fab 转化为全长的IgG-GC121 与SEB有高结合亲和力,与SEA、SEC-1和SED也存在交叉反应。IgG-GC121 可以抑制SEB 诱导的人PBMC IFN-γ 的分泌。预防性使用IgG-GC121 抗体1 h可以保护SEB对小鼠的致死性攻击。研究发现,IgG-GC121 不与STEBVax 结合,可推测该抗体通过阻碍SEB 与MHC II 类分子结合抑制SEB毒性作用。随后,该团队利用同一个噬菌体文库筛选出另一类具有不同结合表位的SEB 人源中和抗体GC132,可以阻断SEB 与TCR 的结合[69]。该抗体与SEB 具有皮纳摩尔结合亲和力,体外毒素中和效果与IgG-GC121相当,在TSS模型中可以保护小鼠免受致死攻击。对GC132 轻链和重链CDR 区采用基于鸟枪同源扫描技术的基因工程抗体亲和力成熟研究,筛选出的GC132a 在以INF-γ 释放量为SEB毒性的衡量试验中,表现出优于亲本抗体250倍的效价。鉴于抗体GC121 和GC132a 分别识别SEB 的MHC II 类分子结合和TCR 结合区,为了探究这两个抗体是否存在协同效应,该团队将单链抗体GC132a融合在IgG-GC121 Fc端,构建的双特异性抗体bsAb-121/132a 也展现了优于亲本的毒素中和效果,但不能与IgG-GC132a 相媲美。该研究中的双特异性抗体建立是一种简单且可有效提高治疗效果的策略,对于不同抗体之间连接方式的优化可能会产生更好的治疗策略。2021 年,Hu 等[70]在人噬菌体抗体库中筛选出3 个抗SEB 人源抗体,LXY8、LXY9 和LXY10,其中LXY8 可有效抑制PBMC 活化和细胞因子释放。在体内小鼠模型中,LXY8剂量依赖性地提高了BALB/c小鼠的存活率。进一步研究发现,LXY8 与TCR 竞争性结合SEB,其中SEB176~179是LXY8结合地关键氨基酸位点。
此外,还有一些抗体筛选方法制备得到的SEs抗体也具有良好的中和性能。2014年,Sully等[71]将SEB 的人-鼠嵌合抗体19F1 转化到根癌农杆菌中,制备出具有良好中和能力的植物来源MAb c19F1。Verreault等[72]研究了具有不同结合表位的IgG-GC121和c19F1在恒河猴气溶胶SEB中毒模型中的作用。所有实验组动物均存活,而对照组动物在暴露30~48 h后死亡。2020年,Liu等[73]利用流式筛选出一株人源SEB 抗体M0313,该抗体可以有效抑制SEB诱导的小鼠淋巴细胞和人PBMC细胞因子IL-2、IL-6、INF-γ和TNF-α的释放,并且在金黄色葡萄球菌引起的小鼠败血症模型中产生了较好的保护作用。在致死剂量MRSA252 攻击前24 h 或攻击后1 h 给予小鼠抗体M0313,可使小鼠分别获得100%或50%的保护率(空白对照死亡率为20%)。注射抗体M0313 的小鼠中有77.8% 在JN064(一株表达SEA、SEC、SED、SEE 的金黄色葡萄球菌)诱导的败血症中存活,33.3%在JN028(一株表达SEB的金黄色葡萄球菌)诱导的败血症中存活(空白对照死亡率均为10%)。其中,SEB85~102是M0313发挥关键作用的免疫优势表位。为了解决抗体治疗过程中使用量大的问题,Kroetsch 等[74]在Xia 等[18]研究基础上,利用酵母展示技术以及合理设计和定向进化MAb 3E2,筛选制备出pH 依赖性抗体L2、L6 和L6.R。这些抗体在中性条件下与SEB 结合亲和力高,在酸性条件下与SEB 结合亲和力低,与亲本抗体3E2 相比,显著降低了SEB的循环半衰期。
5 挑战与展望
SEs毒性作用主要包括超抗原活性和胃肠道活性,其中超抗原活性引起的细胞因子紊乱最终导致TSS,胃肠道活性的致病机制目前尚无定论[29]。TCR-SEs-MHC II 类分子三元复合物是SEs 发挥超抗原活性的必需结构,因此目前针对SEs中毒的免疫治疗性研究大多围绕该三元复合物的形成和破坏。研究表明,主动免疫疗法所使用的疫苗可刺激机体免疫系统对可能的中毒产生较强的抵抗反应;被动免疫疗法所使用的抗体特异性强、体内稳定性高,可快速中和体内已有的毒素蛋白。这些研究表明,免疫治疗策略在SEs中毒临床治疗中可发挥巨大潜力。然而尽管早在20 世纪80 年代就已经有SEs中毒的免疫治疗策略,但是迄今为止仍然没有相关疫苗和抗体获得审批。由Chen 等[47]研发的SEB疫苗STEBVax是唯一进入临床试验阶段的SEs候选疫苗,于2015年完成临床I期试验。目前尚无SEs中和抗体进入临床试验阶段。
限制免疫治疗策略发展的因素有以下几点。a. 在主动免疫疗法中,疫苗主要包括传统的灭活疫苗、减毒活疫苗疫苗,以及基因工程亚单位疫苗、重组载体疫苗、核酸疫苗、合成肽疫苗等新型疫苗。已报道的SEs疫苗主要是围绕制备超抗原活性缺失、但仍保留关键作用位点免疫原性的减毒疫苗和基因工程亚单位疫苗。这些疫苗在动物体内都能诱导中和抗体产生,但同时也能诱导非中和抗体产生。疫苗研究过程中发现,经过免疫的机体再次遇到抗原时,非中和抗体与中和抗体和抗原的竞争性结合可能削弱疫苗的保护作用[76]。此外,一种成功的疫苗从研究阶段到临床应用阶段面临大量的挑战,包括抗原和佐剂对于人体的安全性威胁,疫苗设计和生产过程中的高成本需求,疫苗生产、运输、储存过程中的稳定性要求等[77]。b. 在被动免疫疗法中,尽管SEs发挥毒性作用的位点已基本清晰,但高效精准制备针对这些位点的特异性中和抗体仍然十分困难。目前SEs中和抗体的制备技术主要包括传统的杂交瘤技术和噬菌体展示技术。杂交瘤技术融合、筛选效率低,且获得的鼠源MAbs在人体内易被免疫系统识别并清除,因而制备的MAb 需要额外复杂的人源化改造[78]。噬菌体展示技术中的原核表达经常造成抗体蛋白难以正确折叠且轻重链随机配对,筛选到的抗体通常亲和力不高,往往还需体外进化[79]。此外,已报道的SEs中和抗体均是通过盲筛制备,难以得到针对毒素关键毒性作用位点的高亲和力中和抗体。
针对上述制约因素,可从以下3个方面寻找突破。a. 合成肽疫苗是将抗原中已知的或经预测得到的表位氨基酸序列,通过化学合成技术制备的疫苗,其安全性高、制备方式简单[80]。利用已报道的TCR-SEs-MHC II 类分子三元复合物晶体结构或计算机表位预测技术筛选三元复合物相互作用时发挥关键作用的SEs表位氨基酸序列,有望构建仅诱导中和抗体产生的SEs 合成肽疫苗。SEs 的关键毒性作用位点表位氨基酸合成肽在被动免疫疗法抗体制备过程中也可用于筛选特异性SEs 中和抗体。b. 单B细胞抗体制备技术保留了重链和轻链可变区的天然配对,不仅充分保留B细胞多样性,且具有效率高、时间短的特点,有望筛选出全新的鼠源或人源SEs 中和抗体[79]。c. 传统抗体是异源四聚体,稳定性和异源性高。纳米抗体是在骆驼科及鲨鱼科动物血清中大量存在的一种天然轻链缺失的抗体,其免疫原性低、人源化简单、稳定性高、亲和力高且可识别隐藏抗原表位,在毒素中和过程中有很大的潜在应用价值[81]。
SEB作为一种恐怖战剂通常为气雾状,被人体吸入后造成多器官损伤,严重者可导致休克或死亡[25-26],因此口鼻接种疫苗以及黏膜抗体IgA 在SEs中毒的免疫治疗中十分重要。然而目前为止关于黏膜免疫接种途径SEs疫苗及IgA类中和抗体研究较少,预防及治疗呼吸道感染SEs 的效果有限。SEs的黏膜免疫研究在消除公共卫生安全威胁上有很大前景。
