阿尔茨海默病中的淀粉样蛋白*
2023-08-14储超扬单江晖王清娟李丽萍
肖 彪 储超扬 单江晖 王清娟 沈 巍 谢 凯** 李丽萍
(1)宁波大学医学院附属医院康复科,宁波 315211;2)宁波大学医学部生理与药理学科,宁波 315211;3)浙江省戒毒研究重点实验室,宁波 315010)
阿尔茨海默病(Alzheimer’s disease,AD)是以进行性记忆衰退和认知功能障碍为主要特征的神经退行性疾病[1]。据世界阿尔茨海默病报告指出,目前全世界范围内痴呆人数约有4 700 万,其中约2/3 的人患有AD[2]。全球每3 秒钟就有1 例痴呆患者确诊为AD,到2050 年全球AD 患者人数将上升至1.31亿[3]。AD患者平均寿命为8~10年,随着病情进行性加重,病人经受精神和肉体双重折磨[4]。随着社会人口老龄化进一步加剧,AD患者的数量也将激增,给社会的医疗卫生系统带来严峻挑战。然而AD 治疗进展迟缓,目前临床用于治疗AD 患者的药物主要为改善认知症状的药物如胆碱酯酶抑制剂(多奈哌齐、利斯的明)、N-甲基-D-天冬氨酸(N-methyl-D-aspartate,NMDA)受体拮抗剂(盐酸美金刚)和控制精神行为症状的药物如非典型抗精神病药、5-羟色胺类药,但它们仅在一定程度上缓解临床症状,无法阻止或逆转AD 病理进程。β淀粉样蛋白(amyloid β-protein,Aβ)的形成是AD典型病理特征之一,针对Aβ 的免疫治疗如Solanezumab 免疫疗法在临床试验就已夭折,让人们重新审视Aβ 的作用。因此本文将从Aβ 的产生、发展,以及在AD中的作用和治疗情况等方面进行综述,以期全面认识Aβ,为AD 治疗提供新的思路。
1 淀粉样蛋白级联假说
1.1 淀粉样蛋白级联假说的提出
过去许多科学家尝试对AD 发病机制进行解释,最先受到学界大多数人认可的是淀粉样蛋白级联假说(amyloid cascade hypothesis,ACH)。对于该假说的设想,始于1984 年Glenner 从AD 患者的脑膜血管中发现了“β 淀粉样蛋白”,并进行了部分测序,发现其序列与淀粉样斑块中Aβ 相同。由于在唐氏综合征(Down syndrome,DS)中也发现了同样序列的蛋白质,通过对它们基因序列分析,最终在21 号染色体上发现,淀粉样前体蛋白(amyloid precursor protein,APP)的变体与Aβ 的沉积相关[5]。在家族性AD(family Alzheimer’s disease,FAD)患者遗传基因中发现APP 基因突变,为ACH的提出奠定了基础。1992年首次详细、完整地指出在家族性、早发性AD 患者中,由于APP 基因的突变导致Aβ 大量沉积,使神经元内的Ca2+浓度升高,而Ca2+又参与调控Tau蛋白磷酸化,Tau蛋白的过度磷酸化造成了神经纤维缠结和神经元死亡,最后患者逐渐出现认知症状,并发展为痴呆[6-7]。
1.2 淀粉样蛋白级联假说的发展
随着研究的深入,ACH的细节被进一步补充,如在FAD 患者中还存在早老素基因Presenilin-1(PS1)和Presenilin-2(PS2)的突变,该突变可通过影响γ 分泌酶(γ-secretase)使Aβ 产生增加而导致其大量沉积。在散发性AD(sporadic Alzheimer’s disease,SAD)患者中并未发现APP基因突变或调控γ分泌酶的相关基因突变,而年龄或携带载脂蛋白E (apolipoprotein E, APOE) ε4 等位基因(APOE4)是SAD主要的风险因素[8]。APP经裂解酶依次裂解后产生不同肽链长度的Aβ 单体,如Aβ37、Aβ38、Aβ40、Aβ42、Aβ16-22、Aβ4-x等[9-11],其中Aβ42是主要的病理性蛋白,它与AD患者脑内神经元的丢失和认知症状的出现直接相关[12]。单个Aβ 肽也称为Aβ 单体,非常不稳定且极其容易聚集,Aβ 单体在非极性溶剂中呈α 螺旋结构,而在水溶液中则变成β折叠(β-sheet),β折叠的单体是聚集过程的开始。可溶性Aβ 单体可自发地进一步聚集成不规则的、可溶性的聚集体即Aβ 寡聚体(Aβ oligomers),在寡聚体基础上可聚集成具有高度规则的交叉β 折叠结构的低可溶性Aβ 原纤维(Aβ protofibrils)。Aβ 原纤维进一步聚集可形成不溶性的纤维斑块(fibril plaque)[13-15]。现在的级联假说更倾向于认为脑内Aβ42产生和清除失衡是各种AD 致病因素的根源,Aβ42聚集性沉积成为启动下游级联反应,如Tau蛋白高度磷酸化、神经性炎症等一系列事件发生的诱因,导致神经元的丢失,最终出现痴呆症状[16-19]。
1.3 淀粉样蛋白级联假说面临的挑战
然而ACH 发展至今,仍不能全面解释疾病的发生发展,主要面临以下几点质疑与挑战:
a. 在啮齿类动物的研究中发现,其大脑Aβ 斑块出现的时间与认知症状出现的时间并不一致,即Aβ 的沉积与AD 症状表现的时间及严重程度并未有紧密的联系[20-21]。
b. Aβ 在AD 患者脑中的主要致病形式并不明确。在实验动物和临床病例中均发现,存在脑内出现Aβ 斑块却未有症状的现象[22];有研究表明,FAD是由家族基因突变导致Aβ肽的产生增多引起,而SAD 是由野生型Aβ 折叠不当引起,由Aβ40、Aβ42单体形成的低分子质量、可溶性的寡聚体相对于Aβ40、Aβ42单体或纤维斑块具有更高的神经毒性,在临床试验中也发现减少Aβ 单体产生或不溶性原纤维斑块的沉积并不能改善AD患者的临床症状,甚至出现负面影响[23-25]。
c. Aβ与磷酸化Tau蛋白间的关联并不清晰。在AD患者中,Aβ沉积与纤维缠结、神经毒性程度并未有很强的联系,而高度磷酸化的Tau蛋白却与这些病理现象密切相关;ACH也未阐明Aβ与磷酸化Tau蛋白之间关系,及Aβ是怎样促进Tau蛋白的病理改变[26-27]。
虽然假说面临许多质疑与挑战,但Aβ在AD中扮演着重要的角色毋庸置疑。通过对Aβ在AD患者脑中的形成、发展及变化的深入探究,或将进一步了解其在AD进程中发挥的作用。接下来将从3方面来探究Aβ与AD之间的联系。
2 Aβ在AD中的作用机制
2.1 从淀粉样前体蛋白(APP)到Aβ斑块的形成
AD 患者脑中斑块的形成需经过一系列复杂的病理过程,其中Aβ 清除和产生的稳态在斑块形成过程中发挥着重要的作用[28]。Aβ 的前体是APP[29]。APP为I型跨膜蛋白,广泛表达于中枢神经系统和肝、脂肪等器官或组织,在细胞黏附和神经发育过程中都发挥着重要作用[30]。由于APP 在体内转运和细胞定位的不同,会被不同分泌酶分解(图1)。APP在核糖体中产生后,运输至高尔基体中催化成熟,部分APP 在反式高尔基网络(trans-Golgi network,TGN)中被α 分泌酶(α-secretase)和β分泌酶(β-secretase)分解,且α分泌酶和β分泌酶间存在着竞争关系。剩下未加工的APP 以囊泡出芽的形式运送到细胞膜上,大部分被α分泌酶裂解,小部分被内化,并在核内体(endosome)、溶酶体及蛋白酶体中重新裂解[31-32]。APP被α分泌酶和β 分泌酶裂解后产生胞内结构域(C-terminal field,CTF),CTF会继续被γ分泌酶分解[33]。APP被α分泌酶和γ分泌酶顺序裂解产生P3片段的途径被称为非淀粉样蛋白途径,而APP 被β 分泌酶和γ分泌酶顺序裂解产生Aβ 的途径被称为淀粉样蛋白途径。在生理情况下APP 被不同酶切割产生不同产物,这些产物与神经元不同受体相互作用,构成一个平衡网络,以此来维持大脑稳态;若转运或细胞定位紊乱,会使APP裂解产生Aβ增多,甚至可能会直接导致如神经发育与突触可塑性受损等神经系统功能的异常[34-35]。若Aβ 产生与清除失衡,也会导致大脑中Aβ 沉积。在人体中主要有3 种途径清除过量的Aβ(图1)。其一,Aβ被各种蛋白酶系统清除降解。小胶质细胞和星形胶质细胞的胞外如脑啡肽酶(neprilysin,NEP)、胰岛素降解酶(insulin degrading enzyme,IDE)、血管紧张素转换酶(angiotensin converting enzyme,ACE)和胞内如溶酶体、核内体、泛素-蛋白酶体(ubiquitinproteasome system,UPS) 等均可降解可溶性Aβ[36]。其二,在血脑屏障处,Aβ可经脑内内皮细胞的特殊转运系统进入血液而被清除。其三,在细胞间质中,Aβ 可由胶质淋巴系统和血管周围引流通路进入血液而被清除[37]。若Aβ产生异常增多或清除出现障碍,都导致其在脑内的沉积,沉积的β折叠结构的Aβ 单体可自发聚集成核(或称核种子),形成的核结构像种子一样诱导更多Aβ单体进一步聚集到核中形成原纤维,原纤维通过组装、聚集可形成一系列低聚物,也可进一步形成不溶性斑块[38-39]。Aβ 聚集成斑块是多步骤,依赖于成核的一个过程,核种子的形成是其中的限速步骤,它的形成可加快斑块形成过程[40],其中Aβ42单体是成核的主要分子[41]。脑内Aβ 单体、寡聚体、原纤维、斑块等不同形态在AD发病中发挥怎样的作用还有待探究,何种寡聚形态导致认知下降也不明确。
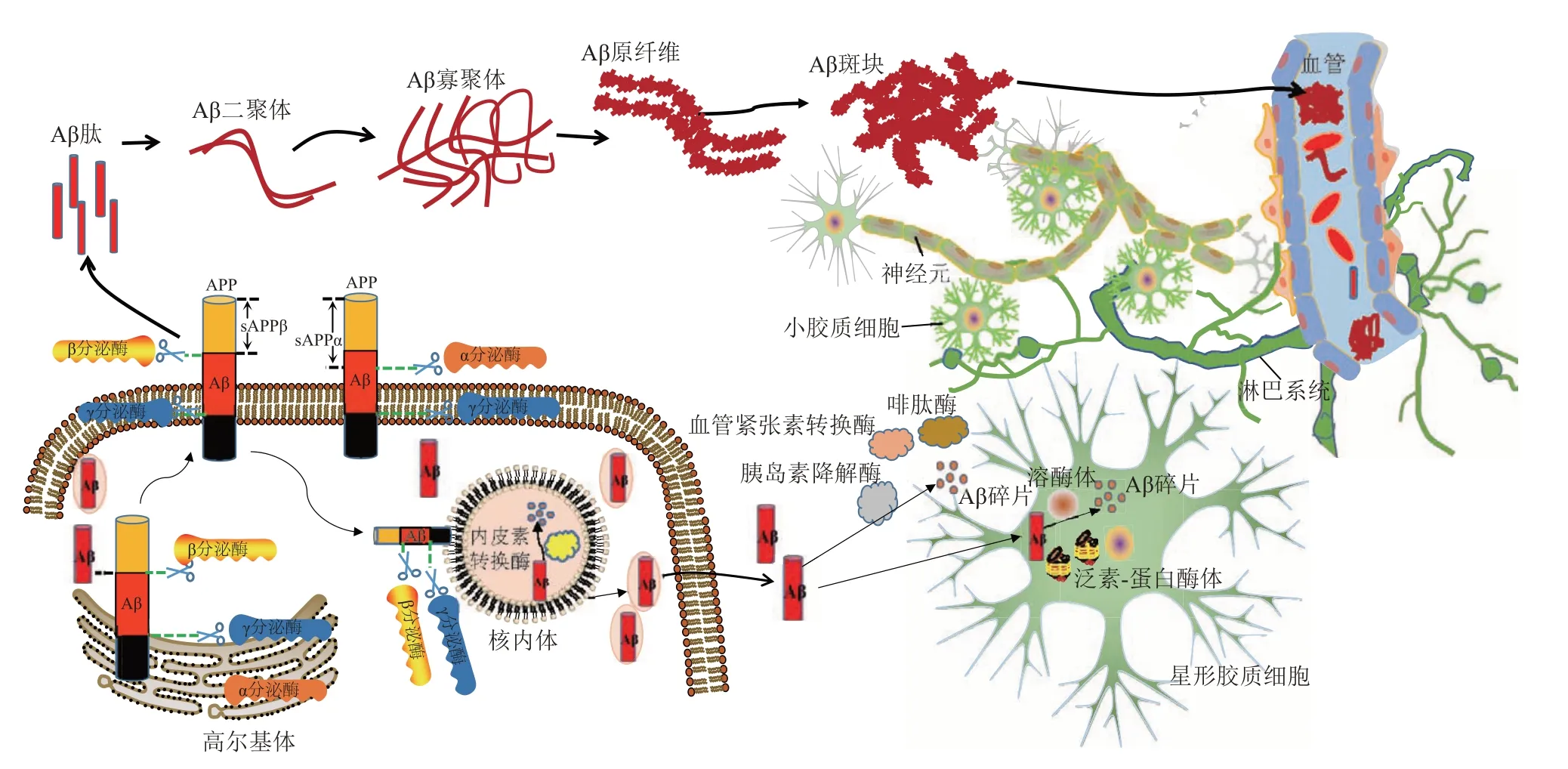
Fig. 1 The process from Aβ peptide production to plaque formation and clearance mechanism图1 Aβ肽的产生到斑块形成过程和清除机制
2.2 Aβ沉积到痴呆症状的出现
Aβ 在诊断或解释部分AD 病理改变上发挥一定的作用[42-43],但斑块和痴呆之间联系及针对清除Aβ治疗手段的夭折,提示ACH已不足以全面描述AD 的症状和发病机制[44]。在Aβ 单体聚集成寡聚体过程中会形成二聚体(两个肽形成)的中间形式。研究发现,Aβ 二聚体可通过抑制突触长时程增强(long-term potentiation,LTP)和降低树突棘的密度,使突触可塑性降低,并损害记忆的形成[45]。Aβ二聚体也可在线粒体中与铜离子相互作用抑制氧化呼吸链中细胞色素C,导致能量供给障碍[44]。Aβ寡聚体可在胞外与不同受体结合产生不同效应,如与膜受体紧密结合阻断膜受体功能、与GM1 神经节苷脂(GM1 ganglioside)结合降低膜稳态,损害神经系统功能[46-47]。此外,Aβ 寡聚体也可在胞内通过过量消耗突触前囊泡或改变突触后修饰位点损害突触功能[48-49]。有研究表明,相对于Aβ 单体而言,Aβ 寡聚体具有更高的神经毒性[50],且寡聚体可通过外泌体在神经元中传播,加速脑内病理进展[51]。然而Aβ沉积并不是导致痴呆症状出现的唯一因素,Aβ 沉积前后还有其他因素共同影响着AD 的进展(图2)。Tau 蛋白是一种微管相关蛋白,主要由N 端、C 端、1 个脯氨酸结构域和1个微管结合结构域构成。微管结合结构域能与不同信号分子相互作用,从而调节轴突和突触功能。正常情况下,Tau蛋白因其特殊回形针样构象而不易自聚集。当病理环境下,Tau蛋白的某些特定基团(如酪氨酸、苏氨酸、丝氨酸残基)被磷酸化,其构象改变而易于聚集,形成神经纤维缠结,沉积于细胞内[52]。可溶性Tau 蛋白出现在疾病早期,导致钙离子信号通路失调以及脑源性神经营养因子 (brain-derived neurotrophic factor,BDNF)水平降低,从而损害突触功能和记忆[26]。不溶性Tau聚集体通过改变膜离子电导、激活电压门控钙通道和NADPH 氧化酶(nicotinamide adenine dinucleotide phosphate oxidase),诱导神经元死亡[53]。此外,神经炎症也可加剧AD进展。小胶质细胞和星形胶质细胞是脑内产生神经炎症的两类主要细胞,它们与神经发生、突触可塑性及大脑的免疫防御相关[19]。小胶质细胞被Aβ寡聚体、磷酸化的Tau 蛋白等诱导因素刺激激活,从“静息态”转变为“激活态”,处于激活态的小胶质细胞因形态发生改变,而吞噬功能受损,导致Aβ 清除减少。激活后的小胶质细胞释放多种炎症因子(如IL-1、TNF-α),加速AD进展[17]。静息型星形胶质细胞可被神经炎症诱导转变反应性星形胶质细胞,反应性星形胶质细胞可上调补体级联基因的表达(如C3),并产生TNF-α、IFN-γ、IL-1β 等促炎因子,导致神经元和少突胶质细胞的死亡[54]。氧化损伤也是AD中常见的损伤因素。氧化损伤是指体内活性氧(ROS)、活性氮(RNS)产生和清除失衡,造成机体生物分子的损伤。在AD中出现DNA和RNA分子的氧化损伤。DNA的氧化损伤可影响启动子的功能,导致基因的转录受损和突变。而RNA氧化损伤会使RNA提前降解,损害蛋白质翻译功能,从而干扰蛋白质合成[55]。NADPH氧化酶4(NADPH oxidase 4,NOX4)是ROS 的主要来源,过表达NOX4可损害星形胶质细胞线粒体电子传递链(electron transport chain,ETC)的功能,抑制线粒体呼吸和ATP产生。形胶质细胞中线粒体氧化呼吸链断裂会增加线粒体ROS 的产生,从而抑制抗氧化,并诱导氧化应激,进一步加重脑内的氧化性损伤[56]。
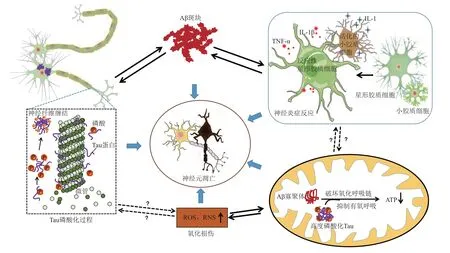
Fig. 2 Multiple factors lead to neuronal damage图2 多因素作用导致神经元损伤
目前关于Aβ、Tau蛋白、神经炎症及氧化损伤在AD发病中具体的关联尚不明确,但它们都可损害神经系统,导致痴呆症状的出现。有研究表明,Aβ 寡聚体主要损伤突触功能,而高度磷酸化Tau蛋白主要损伤轴突功能[57],且它们的可溶形式都出现在AD 早期[58]。总之,Aβ 寡聚体可促进Tau蛋白病理和激活小胶质细胞,诱发神经炎症,而神经炎症又会进一步扩大Aβ和Tau蛋白病理。Aβ寡聚体和Tau 蛋白都会损害线粒体氧化呼吸功能[59-60],增加氧自由基(如ROS)的产生则诱导氧化应激,造成氧化损伤。而氧自由基还可能参与小胶质细胞激活和神经炎症过程[61]。因此,AD中神经系统的损害是一个多因素作用的结果,痴呆症状的出现与Aβ 病理也并不呈单一的线性关系。在看待痴呆症状发生时不应只关注Aβ 单一因素的影响,更需注意多种因素间的相互作用。
2.3 Aβ与家族性AD、散发性AD的相关性
在AD患者中存在FAD和SAD两种疾病亚型,且两者在神经病理方面均表现出脑内及血管壁内Aβ 沉积,但在发病机制及表现形式上却有不同[62]。FAD的发病呈家族聚集性、早发性等特点,主要与APP、PS1、PS2 等基因显性突变有关,这些基因突变几乎直接参与Aβ肽形成过程,使Aβ肽产生增多而加速沉积[63]。Aβ 沉积先于FAD 症状,且Aβ 沉积加速会使症状提前出现,表现出较早的痴呆症状[16]。与FAD 不同,SAD 的发病呈散发性、迟发性特点,其症状出现较FAD 晚,被认为是遗传、代谢、环境等因素共同作用的结果。年龄、APOE基因异常、表观遗传修饰异常被认为是SAD的高风险因素,这些风险因素与Aβ沉积存在联系,但更主要是由于年龄增长、异常表观遗传修饰累积等途径造成体内DNA 片段残缺或丢失,从而影响神经系统正常功能[34,64]。因此,FAD 常表现为单纯的AD 症状,而SAD 多会伴发脑血管病、海马硬化症等。研究发现,Aβ38具有高度聚集性,更易于被成核种子吸引形成斑块。Aβ38在FAD 脑区中积累而在SAD中大多沉积在血管[65]。另有研究发现,SAD 额叶皮质的Aβ38负荷比FAD 多,而Aβ42沉积比FAD 少[66]。这两个相矛盾的研究提示Aβ38或可作为研究FAD 和SAD 关联的关键分子。尽管FAD 与SAD 之间的联系尚不清晰,但从SAD发病来看,Aβ 沉积只是AD 损伤因素之一,其并不足以解释AD的发生及神经系统的损伤。
3 以Aβ为核心的治疗方式
鉴于Aβ 在AD 病理进展中的关键性,涌现许多针对Aβ的治疗方法,主要从抑制Aβ的产生、促进Aβ的清除、抑制Aβ的聚集3方面进行研究。
3.1 抑制Aβ的产生
在Aβ 产生途径的不同阶段进行抑制,表1 列举不同干预手段抑制Aβ 的产生效果。星形胶质细胞通过胆固醇信号调节APP 的转运,再经淀粉样蛋白途径产生Aβ,而低水平胆固醇使APP 经非淀粉样蛋白途径进行降解。在APP 转运阶段,将APP/PS1/Tau(3×Tg-AD)小鼠与SREBP2 基因纯合缺失小鼠杂交可生成星形胶质细胞缺乏胆固醇的AD小鼠模型。在该模型小鼠中,由于APP的转运缺乏胆固醇信号调节,APP从脂质簇中流出,被α分泌酶裂解的机会增加,从而减少Aβ 肽的产生[67-68]。在APP 表达阶段,有研究利用富硒补充剂抑制4月龄的3×Tg-AD小鼠中APP及APP的β位点裂解酶1 (beta-site amyloid precursor protein cleaving enzyme 1,BACE1或β-secretase)的表达,发现调节了AMPK/AKT/mTOR/p70S6K 通路的活性,并促进Aβ 的自噬降解,最终抑制Aβ 的沉积[69]。Sortilin是由sort1基因编码,包含10个蛋白质结构域的空泡蛋白分选受体。Sortilin 的胞内结构域参与调节APP非淀粉样蛋白途径,抑制Aβ产生。在APP 降解阶段,研究发现,在9 月龄APP/PS1(2×Tg-AD)小鼠中敲除sort1基因,可促进淀粉样蛋白途径,使Aβ沉积增加[70]。在APP剪接阶段,通过体内蛋白质表达调节分泌酶BACE1功能。APPSwe突变小鼠细胞质FMR1 相关蛋白2(cytoplasmic FMR1-interacting protein, CYFIP2)表达降低,导致BACE1 及α 钙/钙调素依赖性蛋白激酶II 蛋白增加,造成小鼠脑内Aβ 沉积增加和Tau 蛋白过度磷酸化[71]。过表达Sorting-Nexin-4(SNX4)蛋白或磷酸化真核细胞翻译起始因子4B(eukaryotic initiation factor 4B,eIF4B)均可增加BACE1 的表达,此外SNX4 蛋白还可能通过与BACE1相互作用,阻止BACE1转运到溶酶体,阻断溶酶体对BACE1 降解[72-73]。通过天然或人工合成的化合物调节分泌酶BACE1。淫羊藿次苷II(icariside II,ICA II)是一种天然化合物,来源于淫羊藿属植物的异戊烯基黄酮类成分。研究发现,ICA II 可增加2×Tg-AD 小鼠中ADAM10 表达而抑制BACE1的表达,从而减少Aβ产生,并改善小鼠的空间学习记忆障碍[74-75]。利用烘焙咖啡处理SHSY5Y细胞24 h,发现可以促进BACE1的降解并减少Aβ 沉积[76]。利用20 μmol/L(中剂量)的植物天然提取物白藜芦醇(resveratrol)处理HEK293细胞6~24 h,发现增加APP 产生和Aβ 生成,而利用50 μmol/L(高剂量)白藜芦醇处理却降低β 分泌酶活性,减少Aβ 生成。利用人工合成分子如抗抑郁药物氟西汀(Fluoxetine)对3×Tg-AD 小鼠连续4 个月灌胃给药,发现增强蛋白磷酸酶2A(protein phosphatase 2A, PP2A) 活性和抑制GSK3β 活性,并激活Wnt/β-catenin 信号,最终抑制神经凋亡和Aβ沉积[77]。
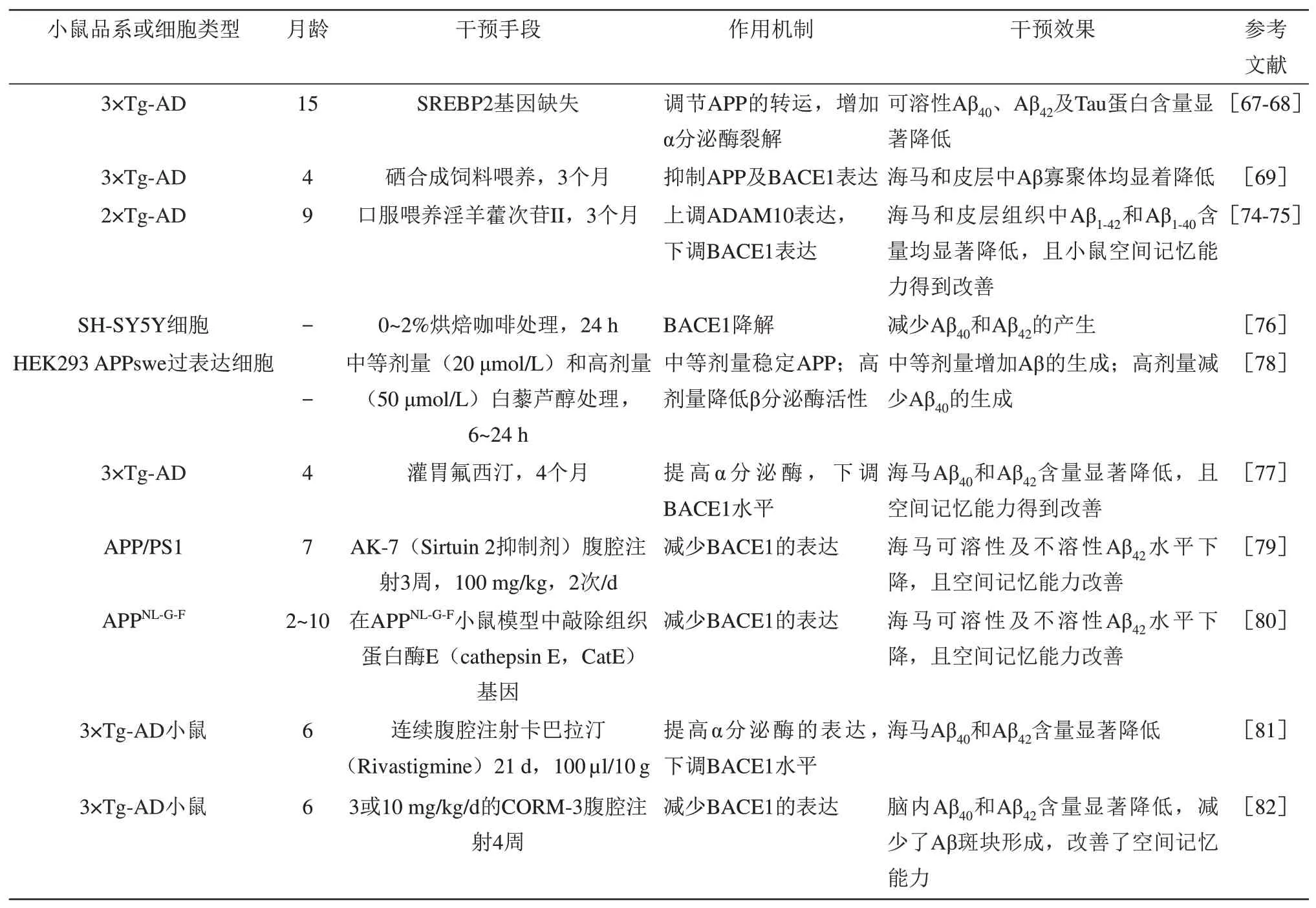
Table 1 Different interventions inhibit the production of amyloid protein in cells or different AD model mice表1 不同干预手段抑制细胞或不同AD模型小鼠中淀粉样蛋白的产生
尽管抑制Aβ 产生的干预方法有许多,但其在AD患者中的临床效果还有待进一步验证。Aβ在正常人群脑部和血液中均有表达,说明其在正常健康人体中可能具有某些生理功能。因此在抑制Aβ 产生时,应注意完全抑制其产生可能对人体产生不利影响。
3.2 促进Aβ的清除
清除Aβ 有直接和间接两种途径。第一代主动免疫试剂AN1792(全长Aβ42与Qs-21 佐剂)可使人体获得对Aβ42的主动免疫,但在一项试验中有6%的受试者产生了T细胞介导的脑膜炎[83],因而第二代的ACC-001和CAD106疫苗,设计为限制性N端抗体,避免了由C端T细胞表位而引起对的脑膜炎[84]。此外,注射外源性抗体如人源性单抗Aducanumab 也可获得对Aβ 的被动免疫。给Tg2576 转基因小鼠尾静脉注射Aducanumab 抗体,抗体可进入大脑与Aβ 结合,并以剂量依赖方式减少可溶和不溶性Aβ 的产生。给轻度AD 或前驱期患者静脉注射Aducanumab 抗体,1 次/月,持续12月,可以减少Aβ的沉积,并减缓认知下降[85]。表2 综述了针对Aβ 清除的临床干预治疗数据。不管是主动免疫还是被动免疫,它们都是直接与体内的Aβ 相互作用而减少Aβ 沉积。与Aβ 直接作用途径不同的是,间接途径通过增加降解酶的表达或提高胶质细胞吞噬能力等方式间接清除Aβ。

Table 2 Clinical interventions and therapeutic effects for Aβ clearance表2 临床上针对Aβ清除的干预措施和治疗效果
在降解Aβ 方面:利用能够稳定和持续表达脑啡肽酶Neprilysin(NEP)的PPAR-sisox9(PBAEPLGA-AgIs-RA-sisox9)纳米制剂转染进神经干细胞,再将神经干细胞移植到9月龄2×Tg-AD小鼠脑部海马区,治疗1、6 个月后小鼠脑部NEP 表达增多,降解Aβ,从而斑块的面积和大小明显降低,并有利于神经干细胞在大脑内存活及分化成熟,从而改善小鼠学习记忆障碍[86];利用贝沙罗汀(Bexarotene)衍生物oAB-14 对8 月龄2×Tg-AD 小鼠进行15 d(急性)和3个月(慢性)的灌胃治疗后,发现oAB-14 提高小胶质细胞吞噬功能和增加IDE 表达,促进Aβ 降解,从而使小鼠海马和皮层中的Aβ40和Aβ42明显降低,斑块沉积面积减小,改善小鼠空间记忆能力[87]。在促进Aβ吞噬和清除方面,CX3CR1是一种在小胶质细胞上表达的趋化因子受体,在CX3CR1 缺乏的2×Tg-AD 小鼠中发现IDE、脑啡肽酶NEP 和基质金属蛋白酶9(matrix metalloproteinase 9,MMP9)显著升高,而脑内Aβ40、Aβ42水平和斑块负荷显著降低,提示CX3CR1 可能参与小胶质细胞的吞噬功能[88]。利用浓度50 或100 mg/L 氧化石墨烯(graphene oxide,GO)处理BV2 小胶质细胞和SH-SY5Y 细胞24 h,可诱导自噬相关蛋白LC3的表达,促进细胞的自噬能力,增强Aβ 的清除[89]。ATG8 家族蛋白(LC3/GABARAPs)与核内体膜结合,可介导小胶质细胞的内化吞噬过程。在5×FAD 小鼠中,缺乏LC3/GABARAPs介导的内吞作用,导致Aβ沉积加重及加速了神经变性[90]。在8~11 月龄2×Tg-AD 小鼠中特异性敲除星形胶质细胞内信号转导子和转录活化子stat-3 (signal transducer and activator of transcription 3),可增强胶质细胞吞噬功能,增强Aβ 的内化和降解,减小斑块负担[91]。除了通过胶质细胞吞噬清除,还可通过类淋巴系统清除。类淋巴系统是一个具有高度极性的脑脊液(cerebrospinal fluid,CSF)转运系统,依赖星形胶质细胞血管侧“终足”(endfeet)上的水通道蛋白4(aquaporin 4,AQP4),通过血管旁(perivascular)和“终足”途径,完成脑脊液和组织液的交换,清除大脑代谢废物,为大脑正常神经活动提供最佳内环境。类淋巴系统功能障碍会造成神经毒性物质的累积,其与生理(如衰老)或病理(如AD等)现象密切相关[92-93]。给8 周龄TgCRND8 AD 小鼠长期抗凝药Dabigatran治疗(每周5 mg/g,治疗22或52 周),发现抗凝药Dabigatran 上调大脑中星形胶质细胞血管侧“终足”上的AQP4表达,从而通过血管旁和“终足”途径促进Aβ 清除,改善小鼠空间记忆能力[94]。此外,敲除12月龄AppNL-F小鼠早期核内体的特异性钠氢交换体6(early endosomespecific sodium hydrogen exchanger 6,EE-NHE6)基因,增强核内体酸化,可提高核内体对Aβ 的清除及恢复囊泡的运输功能,斑块减少了约80%[95]。
综上,主动免疫可以通过短期给药获得长期抗体免疫,成本较小,但由于个体差异性,免疫反应有效性与不良反应较难预测;被动免疫较易控制抗体的浓度与不良反应的产生,但成本较大且给药周期长。通过增加各种Aβ降解酶的表达等间接作用,虽可快速清除或减轻脑内Aβ 的负荷,但这些酶在人体内作用不专一,其底物除Aβ 外,可能还有其他蛋白质,因此可能产生难以预测的副反应或相反的治疗效果。
3.3 抑制Aβ的聚集
目前研究抑制Aβ 聚集的化合物主要有天然化合物和人工合成分子两大类。在天然化合物中,多酚是一种富含芳香族基团的小分子,它们存在于食物或草药提取物中,具有抑制聚集或分解错误折叠聚集体的作用。依据多酚结构不同,可分为黄酮类和非黄酮类[100]。黄酮类多酚如芹菜素,具有稳定Aβ单体、阻止Aβ原纤维化、减少原纤维数量等功能[101]。绿茶中活性成分表没食子儿茶素没食子酸酯(epigallocatechin gallate,EGCG)通过与错误折叠的Aβ直接相互作用及稳定Aβ单体,减少早期阶段Aβ 单体聚集形成低聚物[102]。多肽KLVFFA是Aβ 纤维核心序列肽,可促进Aβ 的纤维化产生。在非黄酮类多酚中,姜黄素可防止多肽KLVFFA堆积,抑制Aβ 寡聚体形成纤维化所需的空间结构[103]。姜黄素还可直接与Aβ 结合并抑制其聚集,从而显著降低了Aβ 斑块负荷[104]。迷迭香酸可与寡聚体Aβ 蛋白直接相互作用从而降低寡聚体稳定性,还可与纤维状Aβ结合抑制其延伸,减弱Aβ神经毒性[105]。此外,疏水和静电相互作用是稳定Aβ聚集体间形成β折叠结构的关键。非多酚类天然化合物芳香族可通过共价/非共价(氢键或电荷)与Aβ疏水区相互作用,亲电碳基可与Aβ的胺和亲核侧链硫醇结合,从而影响一个或多个Aβ 聚集,削弱Aβ 的聚集特性,并可分解成熟的Aβ 原纤维[106]。小檗碱是从中药黄连中分离出的一种季铵生物碱,是一种多靶点的非多酚类天然化合物。小檗碱与Aβ低聚物疏水表面的苯丙氨酸(Phe19)和缬氨酸(Val24)建立疏水键,再与Aβ残基相互作用,抑制Aβ 的纤维化,降低Aβ 神经毒性[107]。在人工合成化合物中,许多新型纳米分子具有抑制Aβ 聚集的作用。原卟啉IX(protoporphyrins IX,PpIX)修饰的氧化碳纳米颗粒是具有多个靶点的新型抗AD 纳米药物,其与聚集超声(3W/cm2,1 MHz)结合作用,可显著降低2×Tg-AD 小鼠中Aβ 的聚集和Tau 蛋白的磷酸化,从而改善小鼠的认知水平[108]。聚(D,L-丙交酯-共-乙交酯)(poly(D,L-lactide-co-glycolide),PLAG)是一种可生物降解的生物相容性聚合物,用于药物输送。绿色合成银纳米颗粒(green synthesis silver nanoparticles,AgNPs)和未结合药物的PLAG纳米颗粒都可通过与Aβ42的疏水结构域相互作用,阻止Aβ42构象向β折叠转变,减少Aβ 聚集。AgNPs 表现出温度依赖性,而PLAG纳米颗粒则表现出特定范围的浓度依赖性[109-110]。天然分子香豆素与N-苄基三唑基团相连,通过人工合成新的香豆素衍生物,可显著降低Aβ 聚集和拮抗小胶质细胞诱导的氧化应激反应,从而起到神经保护作用[111]。另一种香豆素磷酰肼衍生物也可抑制Aβ 聚集[112]。色氨酸-半乳糖缀合物可分解预先形成的Aβ 原纤维[113]。此外,以药物为基础的衍生物,如基于他克林(Tacroline)的强效胆碱抑制剂、他克林与海洋产物PulmonarinB杂合体,可抑制胆碱酯酶和减少Aβ 的聚集[114-115]。苯丙呋喃类似物、 抗真菌药物卡泊芬净(caspofungin,CAS)均可有效抑制Aβ聚集,其中CAS可直接与β折叠部分相互作用,减弱β折叠稳定性[116-117]。此外,一些特殊的小分子化合物,如一种新型带电分子BA-SLOH,具有血脑屏障渗透性和生物相容性,对体内Aβ 斑块进行近红外成像显示,BA-SLOH和Aβ斑块电荷间相互作用,抑制Aβ 自聚集[118]。光活性二氢卟酚e6(chlorin e6,Ce6) 和噻唑烷硫酮(thiazolidinethionone,TZ)修饰的多金属氧酸盐(polyoxometalates,POMs)可对Aβ进行定点修饰,Ce6可选择性破坏Aβ组氨酸残基,而TZ 在赖氨酸16 位点选择性共价修饰Aβ,减少Aβ聚集[119-120]。
尽管针对Aβ的治疗方法很多,但遗憾的是在体外或动物模型上有效的治疗手段,应用在临床上并不一定有效,大部分治疗止步于基础实验研究(表3)。临床治疗的失败,可能有对Aβ致病形式的认识不够清晰、对AD病理不明确及AD动物模型繁杂多样等因素影响。在临床治疗手段上的夭折并不意味着Aβ失去了在AD中的治疗价值。

Table 3 Molecules or compounds that inhibit amyloid-β protein aggregation表3 抑制淀粉样蛋白聚集的分子或化合物
4 AD治疗面临的挑战
虽然已经进行了数十年的基础实验和临床治疗研究,但AD 病理机制没有完全解开,挑战仍存在。目前,AD 的诊断主要采用体液标志物检测、神经影像学检测、神经心理评估及基因检测等方式,但这些诊断方式或具有侵入性或对于AD早期诊断不敏感,而且AD与其他退行性疾病间重叠的临床特征对准确的诊断也提出了挑战[121]。正电子发射断层显像 (positron emission computed tomography,PET)技术提高了AD的诊断特异性。PET检查根据AD相关的标记物质的不同,主要包括 Aβ -PET、 Tau-PET、 氟代脱氧葡萄糖(fluorodeoxyglucose,FDG)-PET 三种,但诊断正确率仅在60%~70%范围[122],且当前中国具备PET检测能力的医院也为数不多。有研究表明,脑脊液病理标志物(如Aβ42、磷酸化Tau 蛋白和总Tau 蛋白)检测联合PET在诊断疾病前驱期、早期等方面具有一定的前景[123]。AD 的发展是一个复杂的、缓慢的过程,Aβ在疾病过程中形态变化的多样性,增加了PET在开发特异性诊断示踪剂上的难度。此外,大多数AD患者在神经元退化和神经可塑性变化之后才开始接受治疗,导致治疗效果不理想,可能与不及时确诊或治疗较晚有关[124],但目前对于AD 早期的概念和确诊的具体时间区段比较模糊,也并未找到与早期相关性强的特异性标志物[125],因此探索早期精准的诊断方式是非常必要的。生物标志物不应只局限于Aβ 或Tau 蛋白,神经炎症贯穿于整个AD病程,神经炎症的变化也值得更多的关注。总之,目前技术无法完全满足早期、快速、特异性的诊断AD的需求。
除了在诊断方面的问题外,AD 动物模型多样化和实用性仍存在较大争议。选择合适的动物模型对于实验的开展及实验结果的临床转化都起着至关重要的作用。在AD研究领域中,转基因动物模型繁多,针对Aβ 斑块和Tau 病理形成的动物模型就有,APP 转基因模型、Tau 转基因模型、PS1 转基因模型、APP/PS1 双转基因模型、APP+PS+Tau 多转基因模型,而且每一种转基因动物模型下又含有多分支的突变体(表4)[126]。在AD 小鼠模型上实验成功的研究结果,却一次次在临床试验中失败,AD 动物模型的适用性也应被重新审视。研究发现,AD 小鼠模型虽然有AD 患者类似的症状和病理,但许多典型病理并未出现,如在APP 小鼠模型中,Aβ 负荷和斑块形成的增加不足以导致Tau蛋白病理变化,此外尽管观察到突触功能障碍和神经元丢失,但随着时间推移也并未观察到明显的脑萎缩现象,这表明AD患者大脑变化的复杂程度在AD 小鼠模型上并不能全面呈现。此外AD 患者大脑并不呈单一的Aβ 相关病理变化,往往还伴随着如DNA/RNA 结合蛋白TDP-43 的聚集和血管疾病为标准的其他病理变化[127]。表达单个FAD突变的转基因小鼠仅可表现出部分病理症状,而需3个或更多的相关基因突变才能复制出人类多数的病理症状,但在AD 患者中少有报道出现多个基因突变[126],提示模拟人类AD并不是简单的病理叠加。另一方面,在临床试验中AD 患者间由于遗传背景、所处社会环境的不同存在明显的个性化特征,而AD 小鼠模型在培养过程中所处环境基本相同,因而在小鼠模型上不能体现环境因素对AD病理变化的影响[127]。AD小鼠与AD患者在药物代谢方面也存在差异性,导致在小鼠上耐受的药物而在人类上并不如此,致使有些药物在临床试验中出现明显副作用而停用。这些问题的存在,提醒人们根据研究目的选择精准合适的动物模型的重要性。
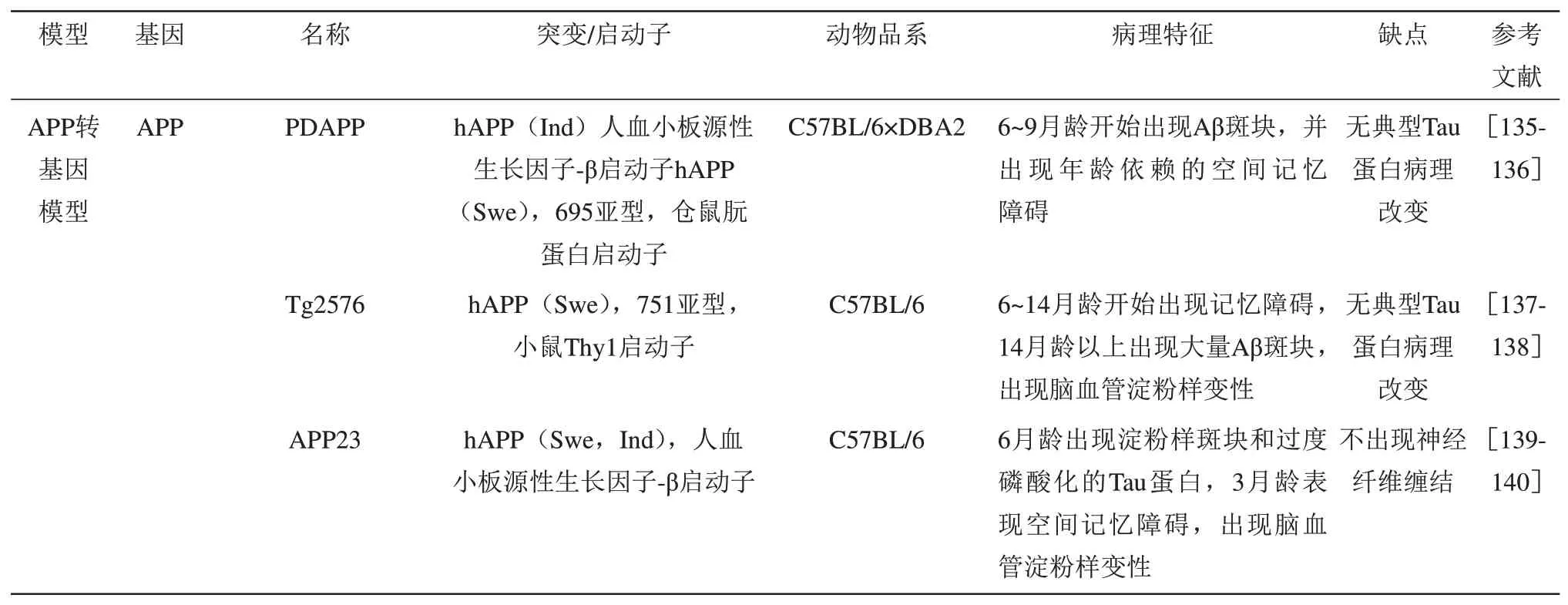
Table 4 Summary table of commonly used AD transgenic animal models表4 常见AD转基因动物模型的归纳表
针对Aβ 的临床药物治疗也面临各种阻碍。首先是针对APP 裂解酶的治疗药物。β 分泌酶(BACE1)和γ 分泌酶都是APP 淀粉样蛋白途径中的关键酶。学术界开展了许多有关两种分泌酶抑制剂的临床试验,但试验结果并不理想。第一代分泌酶抑制剂主要是模仿BACE1 底物序列的拟肽抑制剂(如Verubecestat),存在生物利用度低和穿透血脑屏障差等问题。BACE1 抑制剂在基础研究和临床试验中均发现可减少脑脊液中Aβ 水平,但对于患者认知症状并无明显改善[127]。另外,β 分泌酶和γ分泌酶在体内存在多种底物,如神经调节蛋白1(Neuregulin-1)也是β 分泌酶的底物,它参与了周围神经髓鞘化的过程,γ分泌酶也还可以作用于Notch 蛋白,从而在调节细胞增殖、分化、通信和存活中扮演着重要角色,而使用γ 分泌酶抑制剂(如Semagacestat)后在临床试验中观察到严重的认知恶化[127]。由于这两种分泌酶抑制剂药物的低特异性,在临床试验中产生各种严重的不良反应。α 分泌酶激动剂可减少Aβ 水平和增加sAPPα 片段的产生来发挥神经保护作用,但大多药物处于临床试验前期,试验治疗效果也尚不明确。以抗Aβ 为核心的主动免疫(Aβ 疫苗)与被动免疫(Aβ 抗体)治疗也面临挑战。免疫疗法核心在于诱导特异性自身免疫,在临床试验中免疫治疗方法可使Aβ或斑块水平降低,但均未改善患者的认知情况,反而会加重病情。Aβ 的主动免疫治疗(如AN1792)相较于被动免疫(如Crenezumab)可维持更长的治疗效果,但由于主动免疫药物在AD患者试验中出现脑膜炎、脑水肿、脑内微血管出血等严重免疫副反应,导致这些药物大多在临床试验前期就停止。另外,疫苗在体内难以产生高滴度保护性抗体,并维持长期持续的抗体反应,抗Aβ 抗体在体内的阈值也尚未确定。针对Aβ 单体的被动免疫治疗(如ACC-001)也并未达到改善AD患者认知症状的效果。 针对Aβ 低聚物的抗体(如Aducanumab)产生较小的不良反应,但由于其在临床试验中的治疗效果存在不确定性,对于它们的应用也存在很大的争议。此外,患者在接受免疫治疗还面临免疫耐受性和毒性自身免疫反应的风险,且需要重复给药以维持有效的抗体滴度,其次有些抗体穿透血脑屏障能力很差,因此抗体是否能穿透血脑屏障达到脑部时仍保持有效治疗剂量也是一道难题。
很多靶向Aβ或靶向Aβ生成的分泌酶抑制剂的临床试验都失败了,是否意味着可以否定Aβ 作为靶点的疗效?然而,在这些失败的临床试验中存在着诸多问题。a. 临床试验使用具有安全隐患药物,导致使用药物剂量受限或存在副作用,迫使试验提前停止。b. 试验执行错误。试验前没有预先严格评估大脑中Aβ 水平,以及过早的终止临床试验(患者仅接受12~18 个月治疗),经过短时间治疗可能尚不足以缓解疾病进展。c. 患者选择不理想。纳入的试验者中出现约25%没有AD,或患者开始治疗时间太晚即接受临床治疗时已经达到中晚期AD症状[128]。因此,这些早期的“失败”试验并不能构成反对淀粉样蛋白假说的严格的科学证据。以此放弃抗Aβ 治疗探索将是非常不明智的。尽管其他的靶点(如Tau 蛋白、ApoE4 等)也非常有吸引力,但是研究数据表明这些靶点在减缓AD病理进程中疗效远远低于靶向Aβ[128]。今后如何尝试才能更好证明抗Aβ 治疗是一个合理、有效的靶点?一是可靠的临床前确认。首先基础研究证实某些抗体(药物)有效地降低或中和体内的Aβ 寡聚体或淀粉样蛋白成核种子,然后在临床试验中确认纳入试验者患有AD病理;二是设计一种可靠便捷的生物标记物用于量化和监测AD的病理程度。血浆生物标志物将是一种理想的评估指标,目前比较有前景的标记物是血浆中Aβ42/Aβ40的比例、Tau蛋白的可溶性片段[129]、特异性磷酸化的Tau 蛋白表位等[130-131];结合Aβ-PET、Tau-PET、FDG-PET 等影像学标记物,遴选患AD 的试验者参加临床试验[132];应用血液中某种免疫蛋白水平评估AD患者大脑免疫细胞的激活情况,以此对患者病理进行分层;此外,脑脊液中TREM2 的分裂形式也可成为分类AD 患者的重要依据[133-134]。三是治疗周期至少18~24 个月,且抗Aβ治疗选择一个合适的时间点至关重要,在疾病症状的前期或轻度症状患者延长治疗的时间,产生的治疗效果可能更明显[128]。相信将来会出现更多令人信服的临床证据证明持续降低Aβ 水平可以减少病理性Tau蛋白和改善认知功能衰退。
5 展望
Aβ的发现与研究对ACH的提出有着巨大的贡献,虽然ACH 面临着挑战与质疑,但并不足以将Aβ在AD进展中的作用全盘否定。AD神经病理的形成是一个复杂又长期的过程,Aβ 斑块引起脑内神经元病理改变只是其中的一个诱因,以Aβ 蛋白为核心的临床治疗试验的失利,提示着AD的发生发展是多种因素共同的结果。Aβ 在其中扮演着重要角色,但并不是疾病的全部,它与其他重要因素(如Tau 蛋白、神经炎症、氧化应激等)共同促进了AD的进展。因此,阐明多因素的相互作用是未来AD 研究方向。从Aβ 单体的产生到斑块的形成是一个复杂的过程,且单体、寡聚体、原纤维和斑块各形式之间存在着相互作用,并在疾病发展的各个阶段发挥着不同的作用。因此,针对Aβ 的靶向治疗,要根据不同疾病阶段、不同Aβ 形式选择合适的靶向抗体。现阶段有许多制备AD模型的动物可供研究,但始终缺乏一个能较大程度模拟人脑的疾病模型,导致许多在动物模型上成功的研究却在临床试验中夭折。疾病的早期是治疗的关键时期,但在疾病的早期诊断上仍然面对许多困难。一方面,在AD患者脑部相关病理指标的检测技术方面进展缓慢;另一方面,AD仍未找到较为明确的发病机制,导致在早期AD 定义上的模糊和不准确,相应的早期治疗就难以开展。只有在诊断技术与发病机制的研究上形成合力才有助于AD治疗的迅速发展。此外,不应该只关注到AD 典型病理的本身,病理出现前后脑内微环境的改变可能也是关键的致病因素之一。研究发病前后脑内微环境改变,通过恢复脑内神经细胞适宜的生存环境,或是一种更为有效的治疗方式。
