18p缺失综合征伴肌张力障碍患儿的遗传学分析及文献回顾▲
2023-08-11范美荣宋旭梅于辛酉
范美荣 宋旭梅 于辛酉
(宁夏医科大学总医院1 医学实验中心,2 产前诊断中心,宁夏银川市 750004)
18p缺失综合征也称为18p单体综合征,是由18号染色体短臂全部或部分缺失而引起的疾病,由法国遗传学家Grouchy在1963年首次提出,在活产婴儿中的发病率为1/50 000[1]。该病临床表现个体差异较大,由遗传物质丢失的部位和数量、是否涉及关键基因等决定表型的严重程度,表型包括特殊面容(头围小、上颌突出、眼睑下垂、斜视、眼距过宽、耳位低、鼻梁宽等)、身材矮小、智力发育落后、行为异常和语言运动发育迟缓等,部分患者存在肌张力低下、先天性心脏病、自身免疫性疾病、生殖器官畸形等症状[2]。据统计,有10%~15%的18p缺失综合征患儿存在前脑无裂畸形,导致严重的面部和脑部畸形[3]。
肌张力障碍又称为肌紧张不全或肌张力不全,是一种病理生理机制复杂的运动障碍性疾病,主要的临床症状为不自主、持续性的肌肉收缩,可引起肢体扭曲、重复运动或姿势异常[4]。该病病因复杂,原发性肌张力障碍大多为常染色体显性遗传的单基因遗传病,且可表现出外显率不全的特征[5]。根据不同的分子遗传学病因,原发性肌张力障碍可分为早发型扭转型肌张力障碍(DYT1基因型)、多巴反应性肌张力障碍(DYT5基因型)、混合型肌张力障碍(DYT6基因型)、原发性扭转型肌张力障碍(DYT25基因型)等10个分型[6]。
本研究报告1例18p缺失综合征伴肌张力障碍患儿病例,分析其细胞遗传学、分子遗传学特点及基因型表型的相关性,同时将本病例与近年来国外报告的相关病例进行比较与分析讨论,以提高临床医生对该病的认识。
1 临床资料
患儿,女,7岁,因斜颈2月余,突发口齿不清1周于2020年9月1日入住我院。患儿2个月前出现颈部向右侧歪斜,触之疼痛,头颈无力,患儿家长自行推拿按摩后稍有好转。近1周患儿于感冒后出现口齿不清、说话困难和吞咽困难等症状并呈现进行性加重。患儿系第1胎,父母非近亲结婚,否认遗传病家族史。母亲孕期无异常,孕36周时因胎儿宫内缺氧行剖宫产。患儿存在生长发育落后,2岁时在外院接受过短暂的康复训练(具体不详),无明显效果而停止治疗。2岁5个月可独立行走,至今走路步态不稳,偶有跌倒,4岁时可进行日常的语言交流,精细运动较差。入院时体格检查:身高106 cm(低于同年龄同性别儿童均数的3个标准差水平),体重15 kg(低于同年龄同性别儿童均数的3个标准差水平),神志清,精神反应尚可,无抽搐,四肢活动可;有特殊面容,包括小头畸形、发际线低、短颈、眼距过宽、耳位偏低;四肢肌力5级,轮替动作慢,手握筷子或笔时痉挛,行走时僵硬,不能直线行走;咽反射和双侧膝反射未见异常,双侧巴氏征阴性。头颅MRI及胸部X线片检查未见异常,动态脑电图背景节律慢化,肌电图无异常,血液相关检查及血尿串联质谱分析等均未发现明显异常。按照《韦氏儿童智力量表》评估患儿的IQ为60,判定为轻度智力低下。根据《国际肌张力障碍现象学和分类专家共识》[7]及《肌张力障碍诊断中国专家共识》[8]中的相关标准,判定此患儿存在肌张力障碍。入院后抽取患儿外周静脉血进行染色体G显带核型分析、荧光原位杂交技术(fluorescenceinsituhybridization,FISH)分析及阵列比较基因组杂交技术(array comparative genomic hybridization,aCGH)分析,并抽取其父母外周静脉血行染色体G显带核型分析。染色体G显带核型分析结果:患儿染色体核型为46,XX,del(18)(p11.2),其18号染色体短臂末端存在部分缺失(见图1),其父母染色体核型未见异常。FISH分析(中期分裂相)结果提示患儿只有1条正常的18号染色体,即1个正常的绿色信号存在于长臂末端,1个正常的红色信号存在于短臂末端;另一条18号染色体只有长臂末端标记为绿色信号,短臂末端无红色信号,说明该18号染色体短臂末端存在缺失(见图2);该缺失片段位于18p缺失综合征(OMIM编号:146390)区域,内含GNAL (OMIM编号:139312)、USP14 (OMIM编号:607274)、TYMS(OMIM编号:188350)、TGIF1(OMIM编号:602630)、LAMA1 (OMIM编号:150320)、TWSG1(OMIM编号:605049)、SMCHD1(OMIM编号:614982)等共55个OMIM基因。aCGH分析结果:18号染色体18p11.32p11.21区域存在14.97 Mb的缺失[arr(GRCh37) 18p11.32p11.21(136,227-15,106,305)x1](见图3),涉及18p缺失综合征区域。入院诊断为18p缺失综合征伴肌张力障碍。
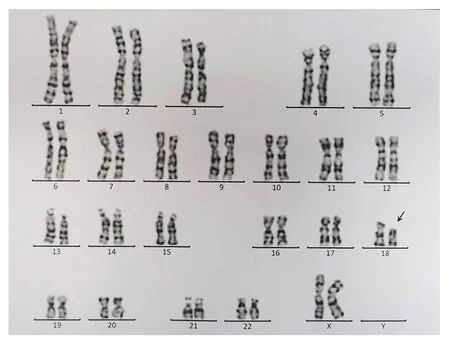
图1 患儿染色体核型图
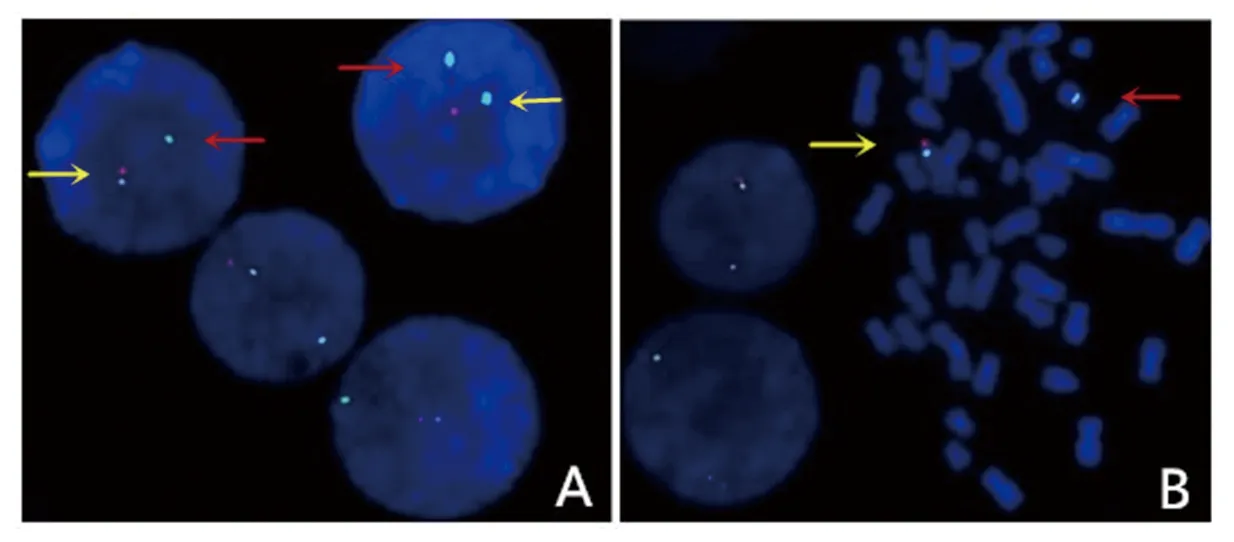
图2 患儿FISH分析结果
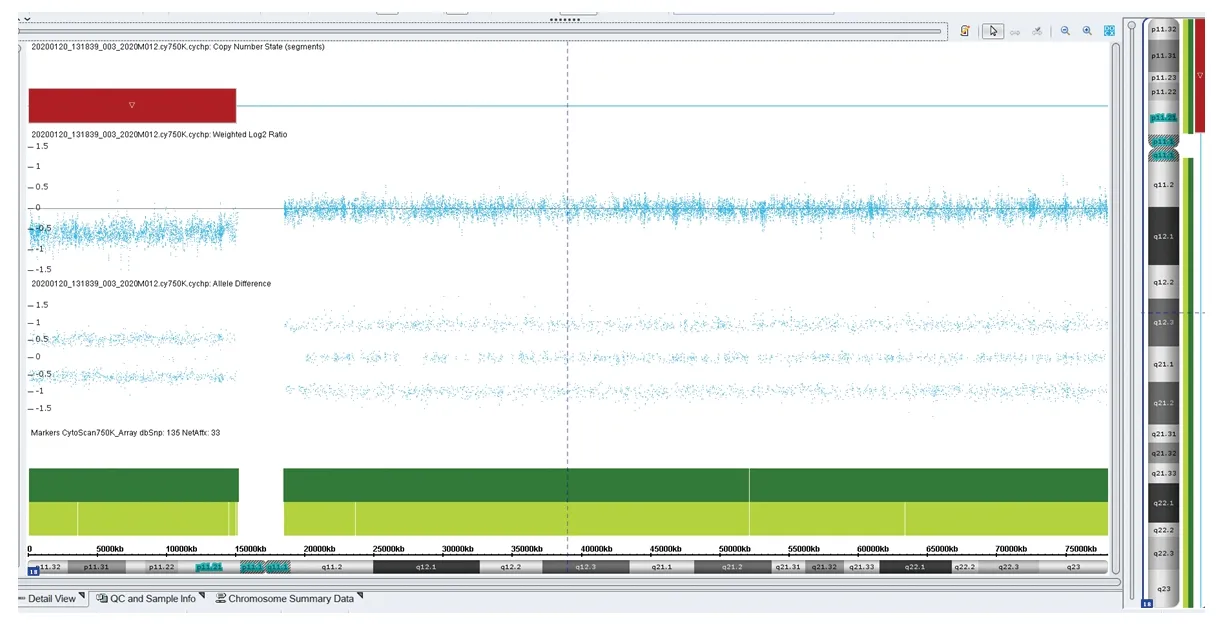
图3 患儿aCGH分析结果
2 讨 论
18p缺失综合征是一种少见的染色体疾病,女性发病率高于男性,该病大部分患儿是由于生殖细胞发育或胚胎早期发育过程中新发的染色体畸变引起,小部分患儿是由于其父母之一为该致病片段的平衡易位携带者,或由于18号环状染色体、18q等臂染色体等造成的18号染色体短臂全部或部分缺失所致[9]。本研究中,患儿父母的染色体核型均正常,故患儿为新发染色体变异。由于18p缺失综合征的临床表型多样,因此相比于其他大多数常见的染色体疾病,该病的诊断更为困难。
本研究aCGH分析结果显示,在18号染色体p11.32p11.21区域存在14.97 Mb的缺失,进一步运用FISH分析技术验证了18号染色体短臂缺失,该缺失片段位于18p缺失综合征(OMIM编号:146390)区域,内含GNAL(OMIM编号:139312)、USP14 (OMIM编号:607274)、 TYMS(OMIM编号:188350)、TGIF1(OMIM编号:602630)、LAMA1 (OMIM编号:150320)、TWSG1(OMIM编号:605049)、SMCHD1(OMIM编号:614982)等共55个OMIM基因。其中缺失的基因表现为单倍剂量不足效应,这些单倍剂量不足基因的拷贝数变异,使基因表达量减少,从而导致一系列症状。TGIF1基因缺失会引起前脑的类维生素A水平增加,从而影响神经轴发育信号通路,是前脑无裂畸形形成的诱因[10]。USP14基因编码的蛋白酶对细胞的生长、迁移、炎症小体活化、激酶活化等过程均起到促进作用[11],该基因缺失会引起生长发育迟缓或瘫痪等症状。TYMS基因异常表达可能增加先天性心脏病的发病风险[12]。SMCHD1基因缺失或突变可导致面肩肱型肌营养不良症2型[13]。本研究中,患儿暂无上述基因缺失的相关表型,分析原因:(1)可能存在表现度差异,患儿年龄尚小,某些症状还未明显表现出来;(2)可能与18p染色体上的许多基因的表型在半合子状态下外显率不全有关;(3)某些基因还受到其他环境因素的影响或基因的调控。
TWSG1基因编码的蛋白是骨形成蛋白的一种结合蛋白,可作为信号转导受体的激动剂参与大脑的发育[14]。Billington等[15]利用基因敲除小鼠模型证实TWSG1基因突变或缺失均是颅面部畸形、前脑无裂畸形和神经管缺陷等表型的危险因素,说明该基因编码的蛋白在大脑发育过程中起到重要的作用。LAMA1基因表达层粘连蛋白,通过负向调节转化生长因子β信号通路,在视网膜和小脑的发育过程中起重要作用,LAMA1基因突变或缺失会导致以小脑共济失调、智力障碍、运动迟缓、语言障碍、近视和视网膜异常为特征的Poretti-Boltshauser综合征[16]。因此TWSG1基因和LAMA1基因的缺失可能是本研究中患儿出现智力障碍的原因之一。
肌张力障碍依据不同的遗传学病因可分为不同的类型,其中GNAL基因缺失可导致原发性扭转型肌张力障碍(DYT25基因型),此分型中大多数患者的首发症状为痉挛性斜颈,痉挛逐渐累及多部位并表现出构音困难[17],本研究中患儿表型符合此分型。Fuchs等[18]于2012年首次选用全外显子测序技术对两个原发性扭转型肌张力障碍家系进行测序,明确了GNAL基因为此病的致病基因。GNAL基因位于18p11.22-p11.21,共有12个外显子,可表达兴奋性G蛋白α亚单位Gαolf,GNAL基因突变或缺失会导致Gαolf合成受阻,并进一步通过影响多巴胺受体腺苷环化酶的合成而引发肌张力障碍[19]。有研究显示,在58例涉及GNAL基因缺失的18p缺失综合征患者中,只有3%的患者出现肌张力障碍相关症状,提示此表型外显率较低[20]。在临床中,大多数18p缺失综合征的患者都是在发病早期得以确诊,但肌张力障碍表型可能出现较晚,因而需要跟踪随访。当18p缺失综合征患者出现更严重的神经或器官疾病表型时,肌张力障碍表型可能会被忽略。另外,因多种疾病可表现出运动障碍及肌张力障碍等症状,因此有这些症状的18p缺失综合征患者很容易被误诊,在遗传咨询时需引起注意。
本研究以“原发性扭转型肌张力障碍(DYT25基因型)”“GNAL”和“18p缺失综合征(18p deletion syndrome)”等词组作为关键词,分别在中国知网、万方数据知识服务平台、PubMed和OMIM等国内外数据库进行检索(检索截止时间为2021年12月),未见国内有因18p缺失综合征(包含GNAL基因缺失)致原发性扭转型肌张力障碍(DYT25基因型)的相关文献报告。将部分国外相关文献报告与本研究进行对比整理,总结如下:(1)既往文献报告的大多数病例于成年期发病,平均发病年龄为31.32岁[21];本病例在7岁发病,较为罕见。(2)大多数患者发病原因相同,但临床症状的严重程度和具体发病年龄等不同,大多数患者都表现出颈部肌张力障碍的症状及因构音困难导致的语言障碍,提示原发性扭转型肌张力障碍表型多样,但又具有一定的典型特点。(3)大多数患者都伴有认知障碍、身材矮小和面部畸形等其他表型,这主要是由于18号染色体短臂缺失区域中还包含其他重要的基因而导致相关表型,因此在临床中遇到肌张力障碍患者特别是伴有智力障碍、面部畸形等症状时,应考虑18p缺失综合征,并及时行分子遗传学检查进一步诊断。
3 小 结
18p缺失综合征是一种罕见的染色体疾病,其临床表型多样,涉及GNAL基因缺失时会出现原发性扭转型肌张力障碍相关症状(痉挛性斜颈、构音困难等),在临床中遇到肌张力障碍患者特别是伴有智力障碍、面部畸形等症状时,应考虑18p缺失综合征,并应用染色体核型分析、FISH分析及aCGH分析等手段进行确诊。
