金属离子(Na+、Ba2+、Ca2+)抑制煤中含氧官能团活性的量子化学研究
2023-08-04何帅印齐金龙周春山唐一博刘洪刚
何帅印,齐金龙,周春山,唐一博,刘洪刚
(1.国家能源集团 神东煤炭集团,陕西 神木 719315;2.太原理工大学,山西 太原 030024)
煤炭是我国重要的基础能源和工业原料,为社会运转和工业生产提供了必要保障。由煤自燃引起的矿井火灾是煤矿常见灾害,且多发于矿井采空区。2001—2022 年我国的煤矿事故数据显示,煤矿火灾事故数占全部类型事故数的比重呈现逐步上升的趋势[1-6]。煤自燃机理的研究是防治煤炭自燃灾害的根本途径,由于煤的结构十分复杂,煤自燃的过程涉及到多种物理化学变化,所以有关煤自燃的研究纷繁复杂。国内外学者提出了各种理论与假说,如细菌导因学说、黄铁矿导因学说、酚羟基作用学说、煤氧复合学说,其中煤氧复合学说由于更为贴近实际而得到了广泛认同。王雪峰等[7-8]、QIAO 等[9]、邓存宝等[10]、WANG 等[11]对钙离子与煤中含氮活性基团形成的配位体的结构进行了全面分析并发现钙离子能够与煤中含氮活性基团形成二配体、三配体及四配体配合物,形成配合物增加了煤中活性结构的稳定性,提高了煤中活性结构的抗氧化性;戴凤威等[12]、邓存宝等[13]发现金属离子与煤分子中的醛基可以形成配合物,从而抑制煤对氧气的吸附来抑制煤的氧化自燃;邓军等[14]通过对不同侧链的相同分子结构的基团进行量子化学研究,发现羟基(-OH)的存在会使-CHO 和-CHOHCH3与氧气的复合反应更容易发生;刘硕等[15]采用透过高度法实验及量子化学计算相结合的方法研究表面活性剂对煤样润湿性的影响效果及机理,发现表面活性剂分子、水分子及煤分子的表面静电势分布共同决定了表面活性剂对煤浸润性的影响;XU 等[16]对低温氧化过程中煤活性的影响以及煤表面官能团的氧化反应进行了研究,发现含氧官能团在煤的低温氧化过程中表现出高活性;葛涛等[17]通过煤中氧结构和碳结构XPS 谱图的拟合分峰和联合解析,发现羟基、羧基和羰基是汾西炼焦煤中的主要含氧官能团,羟基基团含量最高。但相关研究却少见对金属离子与煤炭中含氧官能团相互作用的研究。为此,使用醛基(-CHO)、甲氧基(-OCH3)与苯环结合形成的苯甲醛、苯甲醚作为煤中含氧官能团的2 种简化结构,对煤中含氧官能团在多种金属离子作用下的静电势与反应倾向、结构稳定性、电荷转移进行全面分析。希望能为煤矿阻化剂的应用和煤自燃影响因素的分析提供理论依据。
1 研究内容与方法
1.1 研究内容
根据煤的大分子理论,煤炭氧化过程中,煤分子中含氧官能团、侧链和桥键等活性基团易被氧化,而煤中芳香环的化学性质相对稳定[18],因此选用芳环与煤炭中代表性活性基团组成煤分子的简化结构:苯乙醚、苯乙醛、苯甲醛、苯甲醚、苯甲醇、苯甲酸六种结构进行模拟研究[19],模拟结果表明其中由芳环与醛基(-CHO)和甲氧基(-OCH3)组成的苯甲醛和本甲醚可以与金属离子(Na+、Ca2+、Ba2+)形成配合物,固选择以醛基(-CHO)和甲氧基(-OCH3)与芳环组成的苯甲醛和苯甲醚为研究对象。
从分子角度来看,煤表面由于存在各向异性而导致电荷的不均匀分布,煤分子亲核反应的活性点位位于正点势区域,亲电反应的活性点位位于负电势区域。煤分子的静电势图可以直观的反映金属离子的加入对煤分子亲电反应能力和亲核反应能力的影响;分子轨道理论认为,HOMO 能级越高,越容易失去电子,LOMO 能级越低,越容易得到电子。HOMO 与LUMO 能级差ΔE 的大小反映了电子的轨道跃迁能力,在一定程度上代表了分子参加化学反应能力,即煤分子被氧化的难易程度[20];根据自然键轨道分析,配合物中电子供体轨道和电子受体轨道之间的相互作用稳定化能越大,二者相互作用越强,即电子供体向电子受体提供电子的倾向越大。自然键轨道分析还可得出原子的电荷布局,分子中原子的电荷布局与原子间的相互作用密切相关,配体中各原子电荷大小可以说明配合物的成键方式和电子转移方式。
1.2 研究方法
选用苯环与煤中代表性活性基团醛基(-CHO)、甲氧基(-OCH3)结合所形成的苯甲醛、苯甲醚为煤分子简化结构。使用Gaussian16W 软件,使用密度泛函理论(DFT)的B3LYP 方法,在6-311G(d,p)/LanL2DZ 基组上对含2 种活性基团与3 种金属离子经配合作用形成的配合物进行优化计算,构建出配合物结构,并对分子结构的静电势、体系能量、前线轨道以及自然键理论进行计算分析。煤分子简化结构与配合物的几何构型如图1。

图1 煤分子简化结构与配合物的几何构型Fig.1 Geometric configuration of oxygen-containing groups and complexes
2 结果与讨论
2.1 含氧活性基团和配合物的几何构型
对上文2 种煤分子的简化结构与Na+、Ba2+、Ca2+形成的6 种配合物的产物结构进行假设,再运用量子化学计算方法对这6 种分子结构进行优化计算检验其正确性。使用Gaussian16W 软件,采用密度泛函理论(DFT)的B3LYP 方法,在6-311G(d,p)/LanL2DZ 基组上对2 种氧活性基团及其与3 种金属阳离子配合形成的配合物进行优化计算,得出的可揭示分子几何构型的键长、键角,配合物键长见表1,配合物键角表见表2。

表1 配合物键长Table 1 Bond length of complexes

表2 配合物键角表Table 2 Bond angles of complexes
从表1 可以看出:对于-CHO 与-OCH3,二者与3 种金属离子形成的配位键由长到短排序为:Ba2+>Na+>Ca2+。越短的化学键通常具有更强的稳定性,配合形成3 种配位键的稳定性由强到弱有以下规律Ca2+>Na+>Ba2+。
2.2 静电势与反应倾向分析
含氧结构与配合物的静电势图投影图如图2。总体来看:静电势为负值出现在图中红色区域,说明该分子在这一区域更容易向其他分子或离子贡献电子,即该分子具有亲核性;而静电势为正值出现在图中蓝色区域,说明该分子在这一区域更容易从其他分子或离子获得电子,即该分子具有亲电性[21]。

图2 含氧结构与配合物的静电势投影图Fig.2 Electrostatic potential projections of oxygen-containing groups and complexes
从图2(a)可看出:煤分子中氧原子附近区域呈红色,电势为负;氢原子附近呈蓝色,电势为正。图2(b)~图2(d)中,煤分子简化结构与Na+、Ba2+、Ca2+形成配合物后,氧原子区域由红色变为绿色,电势由负电势向电中性转变;氢原子区域由蓝色变为绿色,电势由正电势向电中性转变;Na+、Ba2+、Ca2+附近区域呈蓝色,正电势增加,表明Na+、Ba2+、Ca2+的加入会降低-CHO 活性基团的亲核反应能力,同时会在会在煤分子中引入1 个位于金属离子区域的亲电反应活性点位。从图2(e)可以看出:煤分子中氧原子和苯环上的碳原子呈红色,电势为负,氢原子附近呈蓝色,电势为正。图2(f)~图2(h)中,Na+、Ba2+、Ca2+加入后,氧原子和苯环上碳原子区域由红色变为绿色,电势由负电势向电中性转变;氢原子附近区域由蓝色变为绿色,电势由正电势向电中性转变;同时Na+、Ba2+、Ca2+附近小部分区域呈现蓝色,电势为正。上述变化表明:金属离子Na+、Ba2+、Ca2+的加入削弱了-CHO 活性基团与-OCH3活性基团的亲电反应能力且使二者失去了亲核反应能力,抑制了2 种基团的活性。
2.3 前线轨道理论与稳定性分析
活性基团与配合物的最高占据轨道与最低占据轨道(HOMO/LOMO)图如图3、图4。前线轨道理论[22]认为,在1 个分子的所有分子轨道中,能量最高的占据轨道(HOMO)和能量最低的非占据轨道(LUMO)对分子的反应和性质起着决定性的作用,对大多数化学反应而言,在满足分子轨道对称性的条件下,反应在1 个反应物的HOMO 与另一反应物的LUMO 能够产生最大重叠位置及方向上发生。

图3 含氧结构与配合物的HOMO 轨道图Fig.3 The highest occupied orbital of oxygen-containing groups and complexes
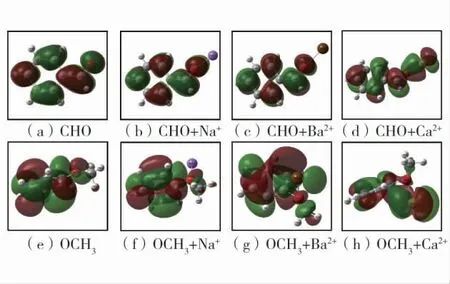
图4 含氧结构与配合物的LOMO 轨道图Fig.4 The lowest occupied orbital of oxygen-containing groups and complexes
从图3(a)、图3((e)中可以看出:-CHO 活性基团的最高占据轨道(HOMO)主要由氧原子和与其相连的碳原子的轨道组成;-OCH3活性基团的最高占据轨道主要由苯环原子和氧原子的轨道组成。从图3(b)~图3(d)中可以看出:-CHO 活性基团在与金属离子形成配合物后,其最高占据轨道主要由苯环上的原子的轨道组成,氧原子对最高占据轨道的贡献大幅降低。从图3(f)~图3(h)中也可以看出,形成配合物后,氧原子对最高占据轨道的贡献有不同程度的降低。从图4 可以看出:-CHO 活性基团和-OCH3活性基团的最低非占据轨道(LOMO)主要由苯环原子和氧原子的轨道组成,形成配合物后,氧原子对最低非占据轨道的贡献下降,表现在图上为氧原子周围红色和绿色区域减少。-CHO 活性基团和-OCH3活性基团主要由氧原子的轨道和苯环上原子的轨道组成,具有较高的化学活性,容易失去电子,可以被氧化发生化学反应进而引发煤自燃。而二者在与3 种金属离子形成配合物后,侧链氧原子对最高占据轨道与最低非占据轨道的贡献均不同程度下降,降低了活性基团的化学活性,提高了活性基团的抗氧化性。活性基团与配合物前沿轨道能量和能级差表见表3。

表3 活性基团与配合物前沿轨道能量和能级差Table 3 Frontier orbital energy and energy level difference eV
从表3 可以看出,-CHO 活性基团和-OCH3活性基团形成配合物后,HOMO 轨道能量绝对值[23]均变大,2 种活性基团与Na+与Ba2+形成的配合物能级差增大,其中Ba2+形成的配合物后能极差增加量最大,说明配合物的形成,增加了煤分子结构的稳定性,使其被氧化的难度增加,且Ba2+的效果最为明显。
2.4 NBO 自然键理论与电荷转移分析
-CHO 活性基团、-OCH3活性基团与Na+、Ca2+、Ba2+形成配合物的部分自然键轨道分析结果见表4、表5。从表4 可以看出:-CHO 活性基团上O 原子的孤对电子与Na+、Ca2+、Ba2+都存在较强的相互作用;其中O(13)原子与Na+的孤对电子的二阶稳定化能为3.12 kcal/mol(1 kcal=4.186 kJ),与Ca2+的孤对电子的二阶稳定化能为7.24 kcal/mol,与Ba2+的孤对电子的二阶稳定化能为1.54 kcal/mol;说明O 原子的孤对电子有向Na+、Ca2+、Ba2+转移的倾向,且孤对电子向Ca2+的转移倾向较大。从表5 可以看出:-OCH3活性基团上O 原子的孤对电子同样对Na+、Ca2+、Ba2+存在较强的相互作用;其中O(12)原子与Na+的孤对电子的二阶稳定化能为2.21 kcal/mol,与Ca2+的孤对电子的二阶稳定化能为5.17 kcal/mol,与Ba2+的孤对电子的二阶稳定化能为0.72 kcal/mol;说明O 原子的孤对电子有向Na+、Ca2+、Ba2+转移的倾向,且孤对电子向Ca2+的转移倾向较大。以上分析说明-CHO 活性基团、-OCH3活性基团与3 种金属离子之间发生了较强的配位作用。
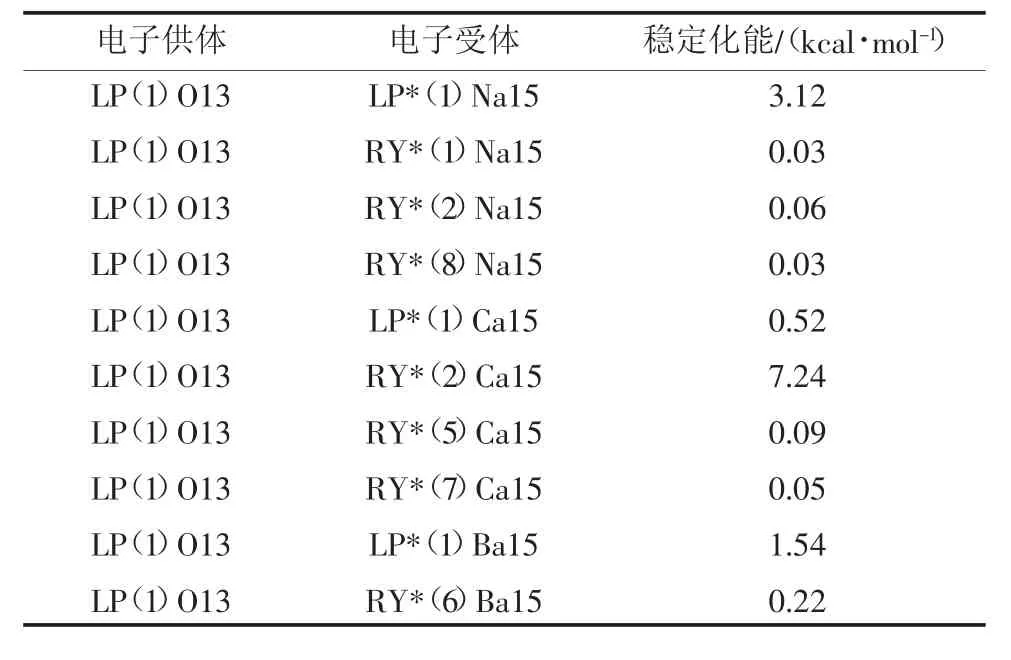
表4 -CHO 形成配合物的自然键轨道分析部分结果汇总Table 4 Natural bond orbital analysis of complexes formed by -CHO

表5 -OCH3 形成配合物的自然键轨道分析部分结果汇总Table 5 Natural bond orbital analysis of complexes formed by -OCH3
根据自然键轨道分析,配合物中各原子电荷见表6。从表6 中可以看出:-CHO、-OCH3活性基团与Na+、Ca2+、Ba2+3 种金属离子形成配合物时,正电荷主要集中在Na+、Ca2+和Ba2+上,带电荷数分别为0.482 41、1.896 40、1.994 61、0.983 65、1.884 25、1.997 29;负电荷则主要集中在-CHO 基团的O(13)上和-OCH3的O(12)上,其余负电荷分布在C 原子上。上述分析说明,Na+、Ca2+、Ba2+3种金属离子与-CHO、-OCH3活性基团上的O 形成了配位键,O 上的电子转移到Na+、Ca2+和Ba2+上;活性基团与金属离子结合后减少了与氧接触的机会,降低了煤自燃的可能性。
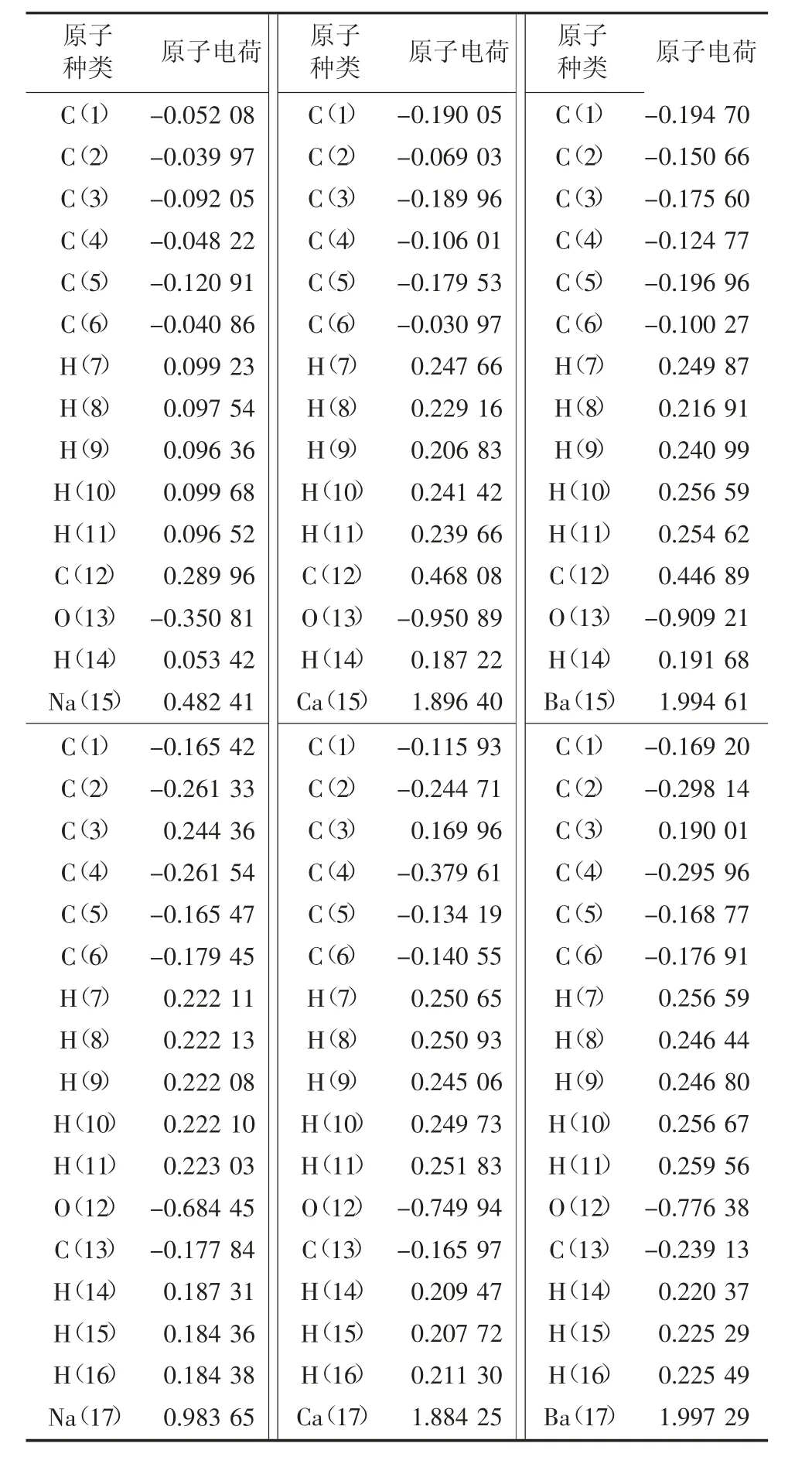
表6 -CHO 、-OCH3 形成配合物的静电荷布局Table 6 Static charge layout of complexes formed by-CHO, -OCH3
3 结 论
1)对于-CHO 与-OCH3活性基团,二者与3 种金属离子形成的配位键由长到短的排序为:Ba2+>Na+>Ca2+;3 种配位键的稳定性由强到弱有以下规律:Ca2+>Na+>Ba2+。
2)煤分子简化结构与Na+、Ba2+、Ca2+形成配合物后,氧原子区域电势由负点势向电中性变化。氢原子区域电势由正电势向电中性变化;Na+、Ba2+、Ca2+的加入削弱了-CHO 活性基团与-OCH3活性基团的亲电反应能力并使二者失去了亲核反应能力。
3)-CHO 活性基团和-OCH3活性基团形成配合物后,二者的HOMO 轨道能量绝对值均不同程度变大,2 种活性基团与Na+与Ba2+形成配合物的能级差(ELOMO-EHOMO)增大,其中Ba2+与-CHO、-OCH3形成配合物的轨道能量绝对值分别为0.231 29 与0.280 46,大于Ca2+、Na+形成配合物的轨道能量绝对值。
4)-CHO、-OCH3活性基团与Na+、Ca2+、Ba2+形成配合物时,正电荷主要集中在Na+、Ca2+和Ba2+上;负电荷则主要集中在-CHO 基团的O(13)上和-OCH3的O(12)上;Na+、Ca2+、Ba2+与-CHO、-OCH3活性基团上的O 形成了配位键,O 上的电子转移到Na+、Ca2+和Ba2+上。
