细胞铁死亡的形态学特征及相关疾病治疗*
2023-06-20赵悄雅常恒瑞常彦忠
赵悄雅 常恒瑞 常彦忠
(1)河北师范大学生命科学学院铁代谢分子生物学研究室,石家庄 050024;2)河北医科大学第三医院脊柱外科,石家庄 050051)
细胞死亡作为整个细胞生命过程的一个环节,在多细胞生物体的生长发育和代谢稳态的维持中起着重要的作用,是生命的基本特征之一。大量研究证实,细胞凋亡(apoptosis)、程序性坏死(necroptosis) 、 自 噬 (autophagy) 、 焦 亡(pyroptosis)等参与多种疾病的发病过程之中,针对单一的途径进行靶向治疗,多数情况下只能有限地缓解疾病。随着研究的不断深入,新的细胞死亡方式不断发现,为全面认识疾病的发生、发展和治疗提供了更加全面的理论依据。铁死亡是一种铁依赖性的受调控的细胞死亡方式,与铁积累和脂质过氧化这两个主要的生化特征有关。谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)是铁死亡的关键调节因子,当其活性受到抑制,细胞的抗氧化能力下降,造成活性氧(reactive oxygen species,ROS)大量堆积时,引发氧化损伤而导致铁死亡。一直以来,研究者对铁死亡、凋亡、自噬、程序性坏死等细胞死亡的分子调控通路十分重视,其形态学上的特征差异有待得到应有的关注。本文重点就铁死亡与其他细胞死亡方式在形态学特征上的区别及其与疾病的关系进行综述,以期更好地理解鉴别铁死亡和其他细胞死亡类型,为临床上选用恰当治疗手段提供帮助。
1 铁死亡
Dolma 等[1]于2003 年筛选出了一种新的化合物Erastin, 能选择性地杀死大鼠肉瘤(rat sarcoma,RAS)基因突变的肿瘤细胞,在其诱导的细胞死亡中没有观察到细胞核的形态变化、DNA 断裂以及活化的Caspase3,Caspase 抑制剂不能阻断Erastin导致的细胞死亡,这与Caspase 抑制剂能够阻断细胞凋亡有着明显区别。2008 年,Yang 等[2]随后发现了可以触发这类细胞死亡的另外两个小分子,RAS-selective lethal 3(RSL3)和RAS-selective lethal 5 (RSL5),并 且 研 究 发 现RSL3具有快速、有效诱导致癌性RAS协同致死的能力,使用铁螯合剂(deferoxamine,DFO)和抗氧化剂(维生素E)能够抑制其诱导的细胞死亡。经过数年深入研究,2012 年,Dixon 等[3]根据Erastin 及RSL3 的作用特点,将这种细胞死亡形式命名为铁死亡(ferroptosis)。铁死亡的发生受多种基因表达和调控,并涉及不同的信号通路,包括氨基酸代谢、铁代谢、脂代谢等代谢途径,与其他细胞死亡形式相比表现出显著的独特性。铁死亡的主要生化特征是铁积累和脂质过氧化。GPX4的活性可以阻止脂质ROS 的积累,铁死亡可通过抑制谷胱甘肽(glutathione,GSH)或GPX4 的生物合成而触发[4]。亚铁离子通过芬顿反应产生大量ROS,导致脂质过氧化。若GPX4活性降低,则细胞抗氧化能力将会下降,无法及时的将过量堆积的ROS清除,过量的ROS 会导致氧化应激反应,引起细胞氧化损伤,进而导致铁死亡[5]。
2 铁死亡与其他形式细胞死亡的形态学特征
铁死亡与其他形式的细胞死亡方式存在多方面不同的表现。在形态学方面,铁死亡与其他类型的细胞死亡相比,存在着较为明显的特征,成为判断铁死亡的重要指标。
研究发现,在存在铁死亡的细胞中,没有出现如程序性坏死和焦亡一样的细胞肿胀、细胞膜上出现空隙进而失去完整性,没有细胞凋亡过程中的染色质凝缩及边缘化、凋亡小体的形成,没有自噬过程中的双膜自噬小泡积累现象。铁死亡主要表现为线粒体形态的显著变化。线粒体形态是判断细胞死亡或存活的一个重要指标,表现在线粒体外膜和折叠成嵴的线粒体内膜的形态变化,研究表明线粒体内膜特别容易受到氧化损伤,因为它的表面积大,靠近产生超氧化物的呼吸链,磷脂中尤其是心磷脂不饱和脂肪酸含量丰富,外膜相对不容易受到氧化损伤[6]。电镜下,细胞启动铁死亡后,线粒体整体表现为萎缩,线粒体膜密度增加、线粒体嵴减少、退化或消失、线粒体膜电位明显降低或消失、外膜破裂等现象,这些是铁死亡重要且独特的形态学特征(图1a)。
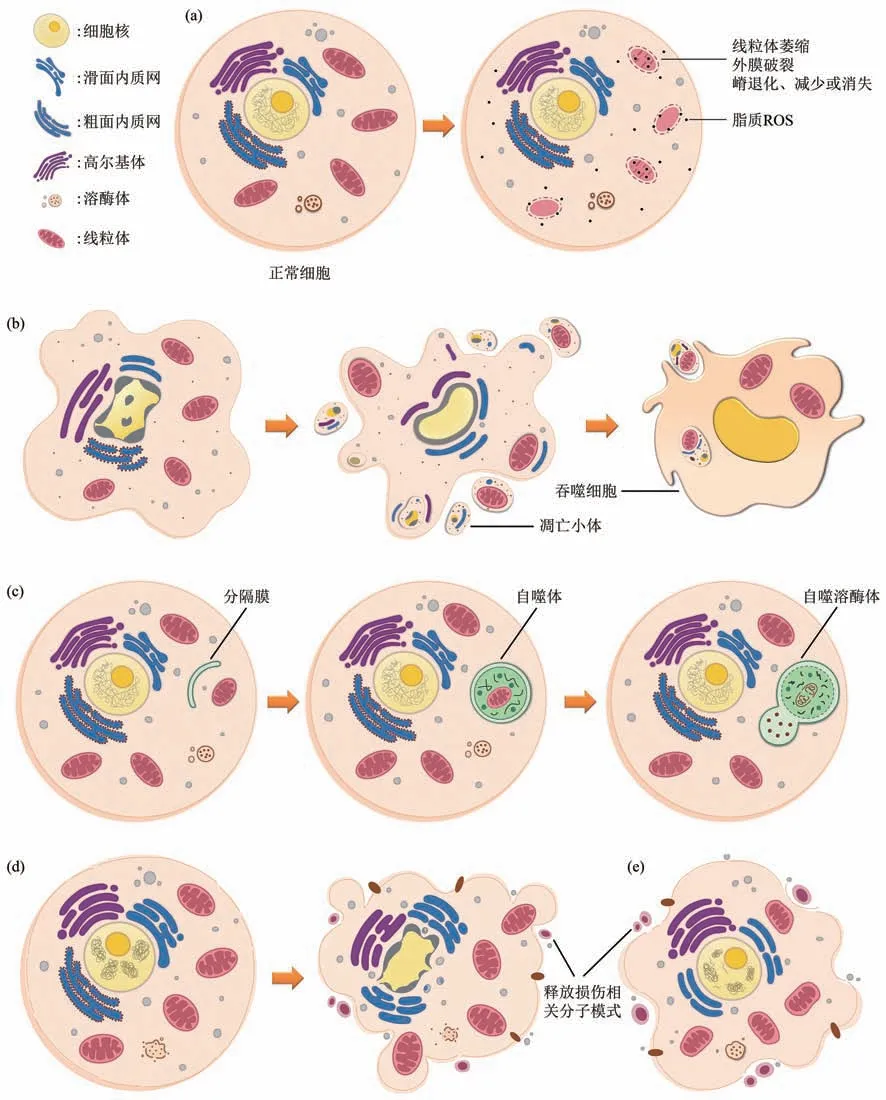
Fig. 1 Morphological characteristics pattern diagram of ferroptosis, apoptosis, autophagy, necroptosis and pyroptosis图1 铁死亡、凋亡、自噬、程序性坏死及焦亡的形态学特征模式图
凋亡是由细胞自身通过调控某种特定的基因或信号通路主动的死亡过程。从形态学上看(图1b),细胞凋亡表现为细胞体积缩小,细胞表面的一些特殊结构,如微绒毛、膜骨架等逐渐消失,与周边细胞分离。细胞骨架解体,细胞质固缩,胞质中线粒体、高尔基体等细胞器凝集,但是细胞器在结构上保持完整,无溶酶体破裂等现象。细胞凋亡典型的形态学变化表现在细胞核浓缩、裂解,核仁消失,核染色质凝集。用末端脱氧核苷酸转移酶介导的dUTP缺口末端标记测定法(TUNEL)对凋亡细胞进行检测,经荧光素等标记后,在荧光显微镜下可以观察到染色体断裂[7]。细胞膜向内折叠包裹胞质以及断裂的染色质片段,包括胞质内紧密排列的完整细胞器,随后逐渐分割形成多个凋亡小体。在细胞凋亡的早期,细胞膜内层的磷脂酰丝氨酸会向外翻转,为吞噬细胞提供识别的信号,从而加速吞噬细胞吞噬清除凋亡小体及凋亡细胞[8]。整个过程中细胞膜完整性保持良好,无细胞内容物外泄,因此不会引起炎症、疾病的发生。对翻转到细胞膜外侧的磷脂酰丝氨酸进行检测,Annexin V染色呈阳性[9]。细胞凋亡过程中,线粒体在形态上不会像铁死亡一样发生明显的变化,但线粒体膜电位会丢失,膜通透性会发生改变,生成多个蛋白质分子构成的孔道,使线粒体中的离子动态流动,向胞质转移,进而诱导细胞凋亡的发生。与凋亡相比,铁死亡细胞的细胞核大小正常且保持完整,没有出现核固缩、裂解的现象,染色质未见发生凝集,也不会形成凋亡小体。但与凋亡相同,细胞膜完整性保持良好,细胞内容物不会渗漏到周围组织。铁死亡细胞的细胞膜虽未见起泡,但细胞膜的磷脂双分子层会发生一定的变化,改变膜的流动性,且膜密度增加[10]。在某些情况下,铁死亡还伴随着细胞的分散和聚集以及自噬体的增加[11]。
自噬是一种通过溶酶体降解内源性底物的生理过程。正常生理条件下,自噬可将细胞内聚集的蛋白质和功能失调的细胞器降解回收再利用,有利于维持细胞内环境稳定,保护细胞免于死亡。然而,如果过度或不受控制的自噬能引起促存活蛋白降解造成细胞死亡,称为“自噬依赖性细胞死亡”, 因此,自噬对于预防或促进细胞死亡具有双重作用[12],这是另一种受调控的细胞死亡形式。在形态学上与凋亡不同的是,发生自噬的细胞,核染色质不凝结,这一特征与铁死亡相同。自噬过程中,细胞被自噬信号诱导激活后,胞浆内双层膜的囊泡包裹了部分或全部胞质、细胞内无用的生物大分子和线粒体、内质网等细胞器碎片,形成自噬小泡或自噬小体[13](图1c)。自噬小体的形成是自噬发生的关键环节,水解的产物可排出胞外,某些营养成分也可被细胞再利用,以维持细胞内循环和代谢稳态[14]。自噬小体形成后,与溶酶体膜结合,变成单层膜,这样,自噬体内容物与溶酶体内容物就会相互混合,形成自噬溶酶体,其内容物随后会被溶酶体中的酸性水解酶类催化降解,最终导致自噬溶酶体破裂水解[15]。通过对铁死亡与自噬特征的研究表明,与自噬类似,铁死亡过程中的氧化应激损伤可诱导溶酶体裂解[16]。与凋亡、铁死亡相似,自噬过程中的细胞膜同样不受影响,保持完整。
程序性坏死,又称坏死性凋亡。程序性坏死具有坏死的典型特征,但其与典型坏死的不同之处是程序性坏死具有可调控的特点,是一种可以被相关分子机制控制的细胞坏死过程[17]。程序性坏死在形态学上整体表现为细胞变圆和肿胀(图1d)。与凋亡相比,程序性坏死中染色质只发生适度凝集,后期细胞核会发生裂解,但不形成凋亡小体,可形成坏死小体,促使细胞死亡的发生。由于细胞质中溶酶体膜被破坏,导致溶酶体水解酶释放到细胞内,破坏胞内其他细胞器,从而出现细胞器肿胀,线粒体功能障碍,线粒体膜通透性增加,线粒体膜电位缺失,细胞肿胀并出现空隙,最终质膜破裂,产生大量细胞碎片,造成细胞死亡。相较于铁死亡、凋亡、自噬等细胞死亡方式的细胞膜保持相对完整不同,程序性坏死过程中,细胞膜上形成许多小孔,破坏细胞膜结构和细胞膜的完整性,导致细胞发生渗透性肿胀,外形变得不规则,细胞内容物等活性物质溢出,引起并放大周围组织的炎症反应。
焦亡是炎症小体引发的一种溶解性程序性细胞死亡[18]。焦亡在形态学方面兼具程序性坏死和细胞凋亡的部分特点(图1e)。细胞发生焦亡时,会在细胞膜上形成大量孔隙,细胞膜完整性丧失,导致细胞失去控制物质进出的能力,进而使细胞膜的通透性增加,细胞内外离子失去平衡,细胞内渗透压升高,细胞器变形,细胞发生渗透性肿胀,细胞内容物会通过细胞膜的孔隙流出,使细胞扁平化,最终导致膜的裂解[19]。这一特征类似于程序性坏死,但与细胞膜结构保持良好完整性的细胞凋亡存在着显著差异。线粒体、溶酶体等细胞器在发生焦亡时也会受到一定的损伤。在质膜破裂前,焦亡的细胞会形成大量的焦亡小体。细胞在发生焦亡过程中还会招募更多的炎症细胞,从而扩大炎症反应[20]。除此之外,细胞膜上形成的孔隙使细胞膜的内侧暴露于外部环境中,因此,经Annexin V 染色后呈阳性[21]。与程序性坏死不同的是,细胞在发生焦亡时也会出现细胞核浓缩、染色质凝缩、DNA片段化等与凋亡相似的特征,因此用TUNEL染色检测焦亡细胞呈阳性。但是焦亡细胞的细胞核完整性保持相对不变,不发生核裂解,这又与凋亡有所区别。研究发现在铁死亡早期,细胞也会与程序性坏死及焦亡一样释放损伤相关分子模式(damage associated molecular patterns,DAMPs),作用于免疫系统,进而引发炎症反应。与Caspase1激活的细胞焦亡不同,铁死亡与程序性坏死都不需要以Caspase作为介质。
由以上分析可知,细胞死亡可分为不受调节的和受调节的死亡两种基本方式,作为受调控的各类细胞死亡方式之间存在着相似之处,但各自之间又有明显的差异(表1)。铁死亡作为近年来受到研究者广泛关注的一种程序性细胞死亡方式,在形态学方面明显不同于凋亡、自噬、程序性坏死、焦亡等死亡形式,其典型的形态学特征表现为细胞体积变小及线粒体结构的显著变化。但是线粒体的一系列改变是否一定引起铁死亡,还有待进一步验证。充分认识铁死亡最基本的形态特征变化及其发生机制具有重要的病理生物学意义。随着不同细胞死亡方式研究的不断深入,进一步分析对比不同细胞死亡方式的形态学特征,探究它们之间的异同,对于辨别疾病过程中不同的细胞死亡形式,准确的病理学鉴别及寻找不同疾病合适的治疗方案具有重要意义。
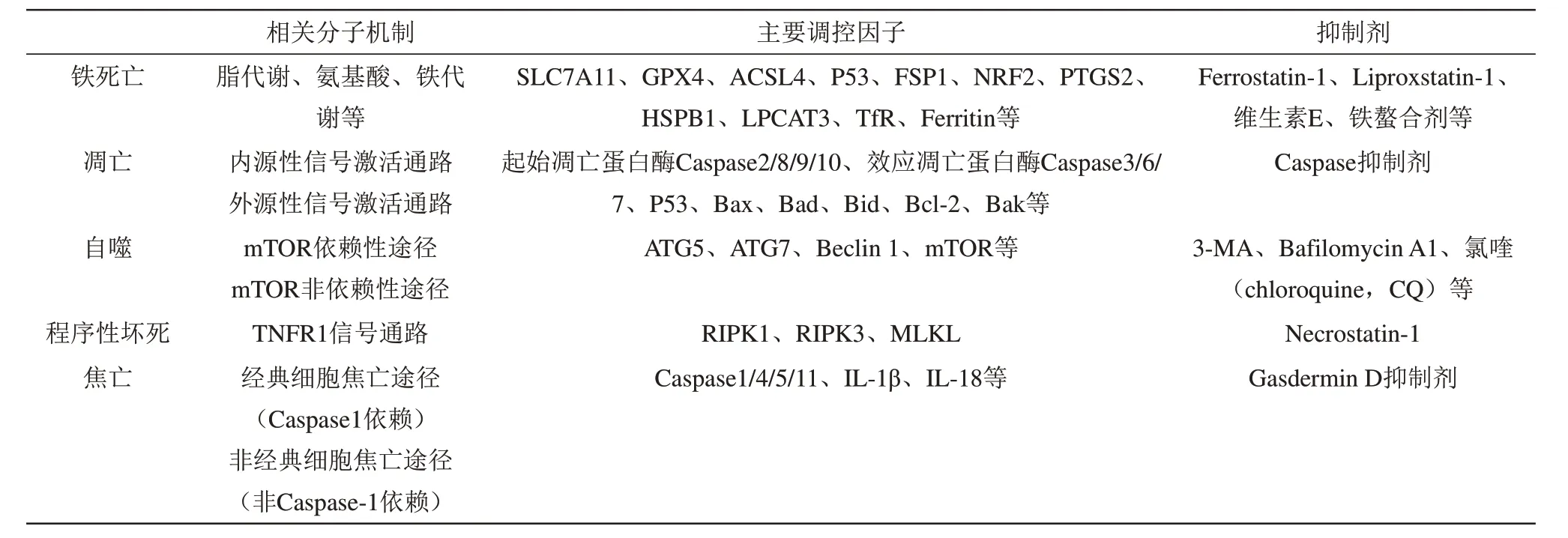
Table 1 Comparison of molecular mechanisms related to ferroptosis, apoptosis, autophagy, necroptosis and pyroptosis表1 铁死亡与凋亡、自噬、程序性坏死及焦亡之间相关分子机制的比较
3 铁死亡与疾病
大量研究表明,阿尔茨海默病(Alzheimer's disease,AD)、帕金森病(Parkinson's disease,PD)等神经退行性疾病、脑卒中、癌症等多种疾病中都有铁死亡的参与,并且铁死亡在疾病的发生和发展中扮演着重要的角色。对铁死亡发生机制及其关键调节因子进一步深入研究,或可为疾病提供重要的治疗靶点。
3.1 铁死亡与脑卒中
脑卒中又称“中风”,是指脑局部血液循环异常引起的神经功能损伤的脑血管疾病。之前对脑卒中引起的脑损伤的研究主要集中在兴奋毒性、炎症、氧化应激以及细胞凋亡等方面[22]。越来越多的证据表明,铁死亡也是脑卒中过程中重要的细胞死亡机制,抑制铁死亡可使脑神经元损伤得到明显缓解[23]。
铁尤其是自由铁池(labile iron pool,LIP)铁的升高和ROS大量积累是造成铁死亡的重要原因,已有研究显示,缺血再灌注造成的脑损伤区域出现铁的明显升高并伴随氧化应激水平的增强[24]。铁在铁死亡的过程中起着重要的作用,减少铁的摄取或增加铁的储存可能会抑制铁死亡[25]。线粒体铁蛋白(mitochondrial ferritin,FtMt)是一种位于线粒体内的铁储存蛋白,在调节细胞铁代谢和保护线粒体免受氧化损伤方面发挥重要作用。研究发现,FtMt 可通过调节细胞质和线粒体间铁平衡,显著抑制Erastin 诱导的细胞内LIP 铁升高及ROS 的积累,FtMt 的过表达可显著降低Erastin 诱导的SH-SY5Y 细胞铁死亡,揭示了FtMt 对Erastin 诱导的铁死亡具有明显的保护作用[25]。缺血性脑卒中患者脑组织中游离铁含量也表现为显著增加[26]。近来在研究FtMt和FtMt相关的铁死亡在脑缺血/再灌注(ischemia-reperfusion,I/R)中的作用时发现,FtMt限制了I/R诱导的铁超载和铁依赖的脂质过氧化,从而抑制了半暗带中细胞的铁死亡;FtMt的缺失促进了I/R诱导的小胶质细胞激活和炎症,通过铁调素(hepcidin)介导的膜铁转运蛋白1(ferroportin1,FPN1)的减少,显著促进游离铁的积累,增加了脂质ROS的生成,加剧大脑I/R中的神经细胞铁死亡[27]。这些结果表明,FtMt 在缺血性脑卒中中起保护作用,FtMt 可能是缺血性卒中的潜在治疗靶点。Tuo 等[28]利用MACO 模型进行研究,发现Tau 蛋白的敲除对I/R 损伤具有神经保护作用,可使这一损伤显著改善,减少铁死亡。表明可以通过抑制Tau蛋白可以缓解铁死亡在缺血性脑卒中中的作用。
脑出血后,红细胞裂解释放血红蛋白,并降解生成大量的游离铁,从而诱发氧化应激损伤,导致神经元死亡加重[29]。研究表明,脑出血中细胞铁死亡可能与血红蛋白诱导的氧化应激损伤有关。Zille等[30]发现脑出血后神经元的死亡具有铁死亡和坏死性凋亡的特征,使用两者的抑制剂可明显防止血红蛋白和血红素诱导的毒性,从而减少神经元的死亡。Li等[31]研究发现,在大脑海马切片培养(OHSCs)模型中,使用ferrostatin-1能够抵抗神经元死亡并降低血红蛋白诱导的铁沉积。另有研究也表明ferrostatin-1 可以减少脂质ROS 的产生,并降低前列腺素内过氧化物合酶2(PTGS2)及其基因产物COX-2 的表达。ferrostatin-1 与其他形式细胞死亡抑制剂的联合应用,可有效地防止在OHSCs和人类诱导的多能干细胞衍生神经元中血红蛋白诱导的细胞死亡[31]。由此可见,ferrostatin-1 可以保护出血性脑卒中铁死亡造成的危害。Zhang 等[32]采用SD 大鼠模型,探讨GPX4 在脑出血后的表达变化及其在继发性脑损伤中的潜在作用和机制,发现GPX4通过调控铁死亡参与了出血性脑卒中后的继发性脑损伤。因此,用特异性抑制剂抑制铁死亡或上调GPX4可能是改善脑出血引起脑损伤的一种潜在策略。
上述分析表明,铁死亡在脑卒中发病机制中有重要的作用及影响。有可能成为脑卒中治疗过程中的一个新靶点。
3.2 铁死亡与神经退行性疾病
大量的研究显示,铁死亡与多种神经退行性疾病,如AD、PD 等发病有关。PD 是一种常见的神经退行性疾病,其病理特点为黑质致密部(substantia nigra pars compacta,SNpc)多巴胺能(dopamine,DA)神经元的缺失[33]。研究表明,PD 患者脑组织中多巴胺能神经元的铁水平明显增加,而铁沉积可以引起多巴胺能神经元的氧化损伤[34]。Zuo 等[35]最近的一项研究表明,百草枯(paraquat,PQ)可以通过铁死亡和铁自噬途径显著诱导多巴胺能神经元的神经毒性。这表明抑制铁死亡可显著改善PQ 引起的神经损伤。当PQ 和特异性铁死亡抑制剂ferrostatin-1 共同作用于SH-SY5Y 神经母细胞瘤细胞时,ferrostatin-1 可通过上调GPX4 和SLC7A11 的表达,抑制脂质ROS的生成铁死亡的发生,从而抑制细胞损伤[35]。铁螯合剂DFO可抑制黑质纹状体退行性变,阻止PD的进展,但DFO 在体内半衰期很短,几乎无法穿透血脑屏障(BBB)。You等[36]设计并制备了一种狂犬病病毒糖蛋白29修饰的去铁胺纳米载药系统,可将DFO 跨血脑屏障送入大脑,能够显著降低脑铁含量抑制细胞调亡,有效治疗MPTP诱导的PD。此外,铁螯合剂去铁酮(DFP)对PD 也可通过抑制铁死亡对神经细胞产生保护作用[37]。AD的主要病理特征是β 淀粉样蛋白(amyloid β,Aβ)斑块和脑内神经原纤维缠结[38]。脑铁代谢紊乱和铁诱导的氧化损伤被认为在AD的发病机制中发挥关键作用[39]。在AD 患者的大脑中,Aβ 对铁有很高的亲和力,加速了铁的积累[40]。铁在大脑中的沉积增加了氧化应激的形成,导致AD患者的大脑中的神经毒性,而铁螯合剂已被证明对AD具有神经保护和恢复神经功能的作用[41]。Xu 等[42]使用APP/PS1 小鼠作为AD 的模型,通过体内证据证明,星形胶质细胞hepcidin 可以通过减少铁沉积来提高APP/PS1 小鼠的空间学习记忆能力,缓解Aβ 聚集引起的神经元细胞死亡。星形胶质细胞特异性过表达hepcidin 可以恢复GPX4 表达水平,并在一定程度上维持了线粒体形态。 铜蓝蛋白(ceruloplasmin,CP)是一种亚铁氧化酶,在调节铁代谢和氧化还原反应中发挥重要作用,经过研究发现CP 对AD 也具有一定的保护作用[43]。另外有研究表明,特异性敲除大脑皮层和海马神经元Gpx4基因的小鼠,其空间学习和记忆功能都有明显的缺陷,并发生海马神经元变性,给予小鼠Liproxstatin-1或维生素 E则可以改善这种情况[44]。由此可见,铁死亡与AD、PD 的发生发展密切相关,并且铁死亡抑制剂在这些神经退行性疾病中显示出巨大的治疗潜力。
除AD、PD 外,亨廷顿病(Huntington's disease,HD)、多发性硬化(multiple sclerosis,MS)等多种神经退行性疾病也已被证实有铁死亡参与。这表明铁死亡在神经退行性疾病中发挥重要作用,对铁死亡的抑制有可能成为神经退行性疾病的潜在治疗靶点。
3.3 铁死亡与癌症
癌症是目前困扰人类健康的一大难题,如何在不损伤正常细胞的情况下,有效地杀死癌细胞,是癌症研究的关键挑战之一。大量研究证实,铁死亡与多种肿瘤的发展如影随形,如在胰腺细胞癌、肾细胞癌、肝细胞癌、乳腺细胞癌、肺细胞癌等癌症研究中,均已发现了细胞铁死亡[45]。与正常细胞相比,为了维持细胞增殖,促进细胞生长,癌细胞对铁的依赖性更强,这种对铁的依赖性也使癌细胞对铁死亡更为敏感[46]。通过外源小分子(如Erastin、索拉非尼)或药物可触发铁死亡[45]。雷公藤红素(celastrol)有良好的抗癌和抑制癌细胞增殖的作用,还可以使临床使用的化疗药物的抗癌作用增强。然而,由于其严重的副作用和较低的生物利用度,使其临床应用受到限制。Liu 等[47]研究发现,铁死亡诱导剂Erastin联合celastrol处理非小细胞肺癌细胞(NSCLC)会增加ROS 的生成,破坏线粒体膜电位,导致线粒体功能障碍,促进线粒体分裂,启动了ATG5/ATG7 依赖的细胞自噬、PINK1/parkin 依赖的有丝分裂以及以HSF1 依赖的方式表达热休克蛋白(HSPs),增加了celastrol 和Erastin 联合使用的抗癌活性。研究发现,干预GPX4表达也成为诱导铁死亡发生的重要手段,如抑制GPX4 产生、促进GPX4 降解或使GPX4 编码基因的缺失,或通过增加可氧化的多不饱和磷脂或干扰铁稳态来破坏脂质代谢平衡,均可使癌细胞对铁死亡更加敏感[48]。此外,通过利用癌细胞与正常细胞之间的某些分子的差异分布,如细胞内铁、GSH、过氧化氢等水平,可以设计药物进行靶向治疗[48]。由此可见铁死亡在癌症治疗及抗癌药物的研发中有着巨大潜力。
总之,以铁死亡为治疗靶点,可从两个方面考虑。一方面,抑制铁死亡可以通过阻断脂质过氧化过程实现。使用铁螯合剂以控制铁水平、使用自由基捕获剂抑制脂质过氧化、通过补硒上调GPX4均可有效抑制铁死亡,延缓病情进展和改善临床症状[49]。另一方面,诱导铁死亡可通过抑制Xc-系统,抑制或促进GPX4降解,减少辅酶Q10,以及通过过氧化物、铁或多不饱和脂肪酸过载,诱导脂质过氧化等方法实现,达到清除特定细胞群(如特定肿瘤类型)的目的[49]。因此,以铁死亡为靶点对疾病的治疗要考虑具体的病理生理特征。
4 总结与展望
铁死亡与其他细胞死亡形式相比,具有独特的形态学特征,主要体现在线粒体形态和功能的显著变化。大量研究表明,脑卒中、神经退行性疾病、癌症等多种疾病中均有铁死亡参与,通过激活或抑制铁死亡可能在相关疾病治疗中发挥重要作用。铁死亡的发现让研究者深入地认识到细胞死亡的复杂性,对于铁死亡,除了已知的信号通路和调节机制以外,是否还存在其他调控铁死亡的因素?铁死亡与其他的细胞生理过程是否存在联系?如何在体内精确诱导铁死亡?对于这些机制和特征进一步解析,有助于更新对疾病的干预方案、寻求更有效治疗相关疾病的特异药物。
