注射用头孢西丁钠杂质谱分析方法影响因素探讨
2023-06-11杨妹妹李玮李姜晖鲁曙光黄鹏
杨妹妹?李玮?李姜晖?鲁曙光?黄鹏

摘要:目的 采用计算机辅助设计试验,考察影响注射用头孢西丁钠有关物质色谱分析方法的主要因素并使目标因子达到最优,对注射用头孢西丁钠杂质谱分析方法进行优化。方法 制备系统适用性混合溶液,确定目标因子。通过Plackett-Burman(PB)设计筛选显著因素;通过中心组合设计(CCD)设计进行优化并确定最佳色谱条件;采用质谱法分析有关物质结构。结果 色谱柱参数、流动相参数和梯度程序等因素均对杂质分离分析有较大影响;在建立的最优色谱条件下,11个潜在杂质均能得到有效分离并被检测。杂质谱分析显示,不同企业产品杂质谱存在差异。结论 使用计算机辅助设计优化色谱分析方法,可实现对潜在杂质的有效分析,为完善注射用头孢西丁钠质量标准和质量控制提供了指导。
关键词:头孢西丁钠;试验设计;杂质谱;有关物质;HPLC
中图分类号:R978.1文献标志码:A
Abstract Objective The computer-aided design experiment was used to investigate the main factors affecting the chromatographic analysis of related substances in cefoxitin sodium for injection and to optimize the objective attributes, and the impurity profile analysis method was optimized. Methods The system suitability solution was prepared, and the objective attributes was determined. The Plackett-Burman (PB) design was used to screen for significant factors, and the Central Composite Design (CCD) design was adopted to determine optimal chromatographic conditions. Mass spectrometry was used to identify the structure of the related substances. Results The column parameters, mobile phases, and the gradient program had great influences on the separation and analysis of impurities. With the optimal chromatographic conditions established, 11 potential impurities were effectively separated and detected. Impurity profiles analysis showed that there were differences in the impurity profiles of samples produced by different enterprises. Conclusion The use of computer-aided design in optimizing chromatographic method can facilitate the effective analysis of potential impurities. The article provided the guidance for cefoxitin sodium for injection standard improvement and quality control of the preparation.
Key words Cefoxitin sodium; Design of experiments; Impurity profiles; Related substances; HPLC
头孢西丁钠(cefoxitin sodium)属于头霉素类抗生素,母核由四元的β-内酰胺环与六元的氢化噻嗪环骈合而成。主要用于敏感细菌所致的呼吸道、泌尿道、骨关节、皮肤软组织及腹内感染等[1-2]。该品种由于制备工艺特殊且自身结构稳定性较差,在生产和储存过程中有关物质容易增加,甚至导致不良反应[3],因此对其杂质谱的分析和控制显得尤为重要。
在建立杂质谱分析方法过程中,如何明确药品中的杂质组成、进而开发适宜的分析方法是首先需要解决的问题[4]。早期对头孢西丁钠有关物质的报道均主要关注于其总量的测定[5-7],注射用头孢西丁钠虽然已被《中国药典》(ChP)、美国药典(USP)、欧洲药典(EP)等收载,但仅EP列出了7个有关物质的结构。近年来,随着研究的深入,对该品种中潜在杂质的结构确认[8-10]和分离分析的报道[11,12]逐渐增加。然而,目前仍缺少系统性建立头孢西丁钠杂质谱色谱分析方法的范例,对影响各杂质分离的主要因素分析也未见报道。本文结合已有的文献,利用混合系统适用性溶液,以难分离杂质对作为色谱分析的目标因子,通过疏水消除模型筛选适用于分离难分离杂质对的最优色谱柱,再利用计算机辅助实验设计方法,寻找头孢西丁钠有关物质分析的最优色谱方法,并考察不同企業样品杂质谱的差异。在此基础上,全面总结注射用头孢西丁钠有关物质分析的关键影响因素,为完善注射用头孢西丁钠的质量控制提供指导。
1 材料与方法
1.1 仪器与试药
岛津LC-20A液相色谱仪;AB Sciex triple quad 5500 质谱仪; SevenExcellence型pH计;电子天平XP205。试验用到的色谱柱信息见表1。
注射用头孢西丁钠样品;头孢西丁杂质对照品:EP杂质B、EP杂质D、EP杂质E、EP杂质F对照品,均由广州佳途科技股份有限公司提供;3-羟甲基头孢西丁(EP杂质A)、7-甲氧基头孢噻吩,均由重庆天地药业有限责任公司提供。
乙腈(色谱纯)为赛默飞世尔科技有限公司产品;甲酸铵、磷酸二氢钾、无水磷酸氢二钠、磷酸、甲酸和30%过氧化氢,均来自国药集团化学试剂有限公司。
1.2 方法
1.2.1 色谱方法的筛选与优化
以ChP(2020年版)、USP(2021年版)和EP(10.0版)收载的头孢西丁钠有关物质分析方法[13-15]作为初始方法。试验过程为:①比较各色谱方法,筛选色谱条件;②确定目标因子,考察难分离物质对的分离度情况;③考察柱参数对目标因子的影响,确定影响最大的柱参数,筛选出合适的色谱柱;④采用Plackett-Burman(PB)设计筛选显著影响色谱分离的主要因素,剔除不显著的因素,以减少后期优化实验的次数;⑤通过单因素实验确定主要影响因素的优化范围;⑥采用中心组合设计(central composite design,CCD)设计进行响应面优化,通过最大化总体期望函数确定最佳色谱条件;⑦样品测定。
1.2.2 强制降解溶液的制备
称取样品0.125 g置于5 mL容量瓶中,用磷酸盐缓冲液溶解并稀释至刻度,作为母液;取1 mL母液置于5 mL容量瓶中,在80℃水浴处理1 h,待冷却后,用磷酸盐缓冲液稀释至刻度,即强制降解溶液1;取1 mL母液置于5 mL容量瓶中,加1 mL 3%H2O2,2 h后加入磷酸盐缓冲液稀释至刻度,即强制降解溶液2。
1.2.3 系统适用性溶液的制备
诸头孢西丁杂质对照品用磷酸缓冲液溶解,分别制成浓度为1 mg/mL的杂质溶液;分别取EP杂质B和3-羟甲基头孢西丁杂质溶液各50 μL、EP杂质D、EP杂质E、EP杂质F和7-甲氧基头孢噻吩杂质溶液各100 μL,与强制降解溶液1和2各300 μL混合,得到混合系统适用性溶液。
1.2.4 质谱分析条件
电喷雾离子源(ESI);正负离子扫描方式,扫描范围m/z 330~800;气帘气压(CUR) 20 psi;碰撞气压(CAD)7 psi;电喷雾电压(IS) 5500 V;雾化气压(GS1)16 pai。
2 结果
2.1 色谱条件初筛与目标因子确定
对不同头孢西丁钠有关物质分析方法的筛选结果显示,USP方法中,主成分保留时间过长,峰形差,且主峰之后无明显杂质峰;ChP方法中检测到的杂质数目有所增加;使用EP方法检测到的杂质数目最多,且各色谱峰的理论板数较高(图1A)。其中,色谱峰1、2、3、4、5、6、7均为强制破坏后增加的杂质峰,峰6为EP杂质A,峰8为EP杂质D、峰9为EP杂质E、峰10为EP杂质F、峰11为EP杂质B、峰12为7-甲氧基头孢噻吩。故在此基础上进行色谱条件的优化,该阶段的目标因子初步选择为:难分离物质对峰3和4的分离度(R3/4);难分离物质对峰8和9的分离度(R8/9);峰4的理论塔板数R4N和峰8的理论塔板数R8N。
2.2 柱参数对目标因子的影响
由于采用EP规定的色谱柱试验结果并不理想,故首先考察色谱柱参数对目标因子的影响。选取11根不同色谱柱进行分析,试验结果见表2。
用Snyder等基于疏水消除模型提出的色谱柱参数表征不同色谱柱的差异[16],将目标因子与柱参数进行多元线性回归,其中C(2.7)由C(2.8)和C(7.0)通过插值法计算得到[17]。方程分别为:
R3/4=54H-109S-12A-15B+15C2.7(1)
R8/9=39H-49S-9A+20B+7C2.7(2)
R4N=341951H-757200S-87834A-252006B+ 31380C2.7(3)
R8N=864088H-1643173S-291179A-337865B-9569C2.7(4)
利用ANOVA对诸线性回归方程进行分析,P值均小于0.05,即因变量-自变量拟合方程(模型)有意义;采用t检验考察色谱柱参数的影响,所得的P值中,柱参数H的显著性均小于0.05,表明疏水性对目标因子的影响最大,提示EP规定的苯基柱可能并非最佳色谱柱类型。选择对诸难分离色谱峰分离最优且柱参数H值较大的ZORBAX Eclipse XDB-C18和ZORBAX SB-C18色谱柱进行后续试验,考察其他因素对目标因子的影响。
2.3 筛选主要影响因素
流动相的pH、浓度、有机相比例、梯度时间、色谱柱类型、柱温、流速等均可能对目标因子产生影响。由于影响因素较多,首先采用Plackett-Burman(PB)设计剔除影响不显著因素。确定优化目标因子为:难分离色谱峰2和峰3的分离度(R2/3)、主峰和峰8的分离度(R主峰/8)、峰8和9的分离度(R8/9)、峰9和10的分离度(R9/10)、峰10和11的分离度(R10/11)和峰12的保留时间(tR12)。影响因素考察范围为,A:梯度开始时有机相的比例5%~12%;B:梯度结束时有机相的比例 20%~35%;C:流动相pH 2~4;D:柱温 30℃~40℃;E:流速0.8~1.0 mL/min;F:甲酸铵浓度0.5~2.0 g/L;G:梯度开始的时间2~10 min;H:梯度结束的时间30~60 min;J:色譜柱类型ZORBAX Eclipse XDB-C18、ZORBAX SB C18。
共进行12组试验考查各因素对目标因子的影响,回归分析结果显示,流动相pH对各目标因子均有显著影响;梯度开始时的乙腈的比例,对除R10/11外的各目标因子均有影响;柱温越低,R10/11越大;梯度结束时间对tR12有一定影响。故对流动相pH、梯度开始时乙腈的比例、梯度结束时间和柱温进行后续优化。
2.4 CCD优化
根据筛选出的主要影响因素,采用ZORBAX Eclipse XDB-C18色谱柱,利用单因素实验确定各影响因素的优化范围。结果表明,流动相pH=3.0时R2/3较低;pH=3.9时,色谱峰8和峰9的位置颠倒,最终设定pH优化范围为3~4。梯度开始时乙腈的比例为3%时,峰1呈融合峰;为9%时,分离度尚可;为12%时,峰1和2未分开,故设定初始乙腈比例的优化范围为2%~12%。梯度結束时间增加有利于分离,兼顾分析时间,设定为50~60 min。预实验表明,低柱温有利于各杂质的分离,为易于控制,设定柱温范围30℃~40℃。
根据单因素实验确定的优化范围,采用CCD继续优化。按随机顺序进行试验,得到对应的响应值(表3)。
试验得到的回归方程分别为:
R2/3=2.6-1.24A+0.99B+0.32C-0.26D+0.28A×B-0.28A×D-0.27B×C+0.48B×D-0.52C×D-0.2A2-0.3B2-0.5C2(5)
R主峰/8=4.26-0.26A+0.33B+0.14C-0.61A2+ 0.36B2+0.19C2;(6)
R8/9=1.2-2B+0.72C (7)
R9/10=3.11+9.292E-003A+0.21B+0.014C+ 0.072D-0.018A×B+0.053A×C-0.071B×C+0.021B× D-0.29A2-0.089B2-0.018D2(8)
R10/11=3.81+0.21A-0.014B-1.25C-0.073D+ 0.054A×B+0.057A×C-0.12B×D+0.054A2-0.51B2+ 0.022D2(9)
tR12=40.37-7.93A-4.05B-1.21C+1.77D-1.01A×B-0.95A×D-2.08A2+B2(10)
诸模型均显著,因素各交互作用的3D图见图2。采用满意度函数法(表4)确定最优化色谱系统,最优色谱图如图1C所示。根据模型预测,该条件下R2/3、R主峰/8、R8/9、R9/10、R10/11和tR12的预测值分别为2.46、3.87、2.76、2.59、3.58和52.29 min,实际测定值为1.741、3.733、3.612、2.645、3.86、54.767 min,提示模型预测准确。与ChP(图1B)和EP(图1A)方法相比,优化的色谱方法更优,各杂质峰均达到理想的分离水平。
2.5 实际样品测定
选择5个不同生产厂家的市售注射用头孢西丁钠(编号为A、B、C、D、E)进行测定(图3):仅厂家A的产品中检出峰5;5个厂家的产品中均未检出峰7(氧化降解产物)和峰12;厂家C的产品中未检出峰10;厂家B的产品中未检出峰11;其余杂质峰在各厂家产品中均有检出。厂家C产品中峰8的含量最大(0.28%);厂家D产品中峰6的含量最大(0.26%),且其杂质总量最高(0.99%)。
2.6 质谱分析
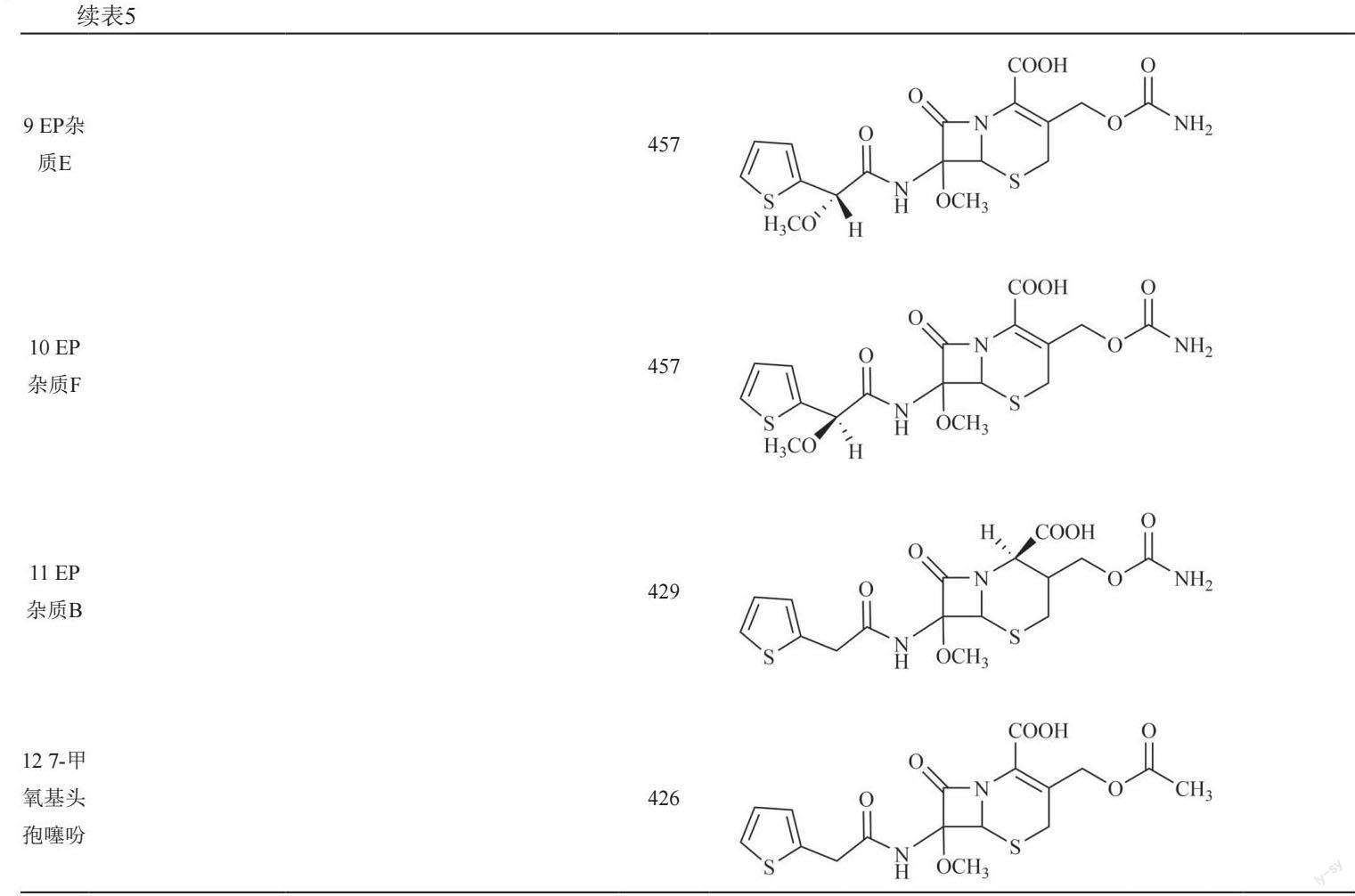
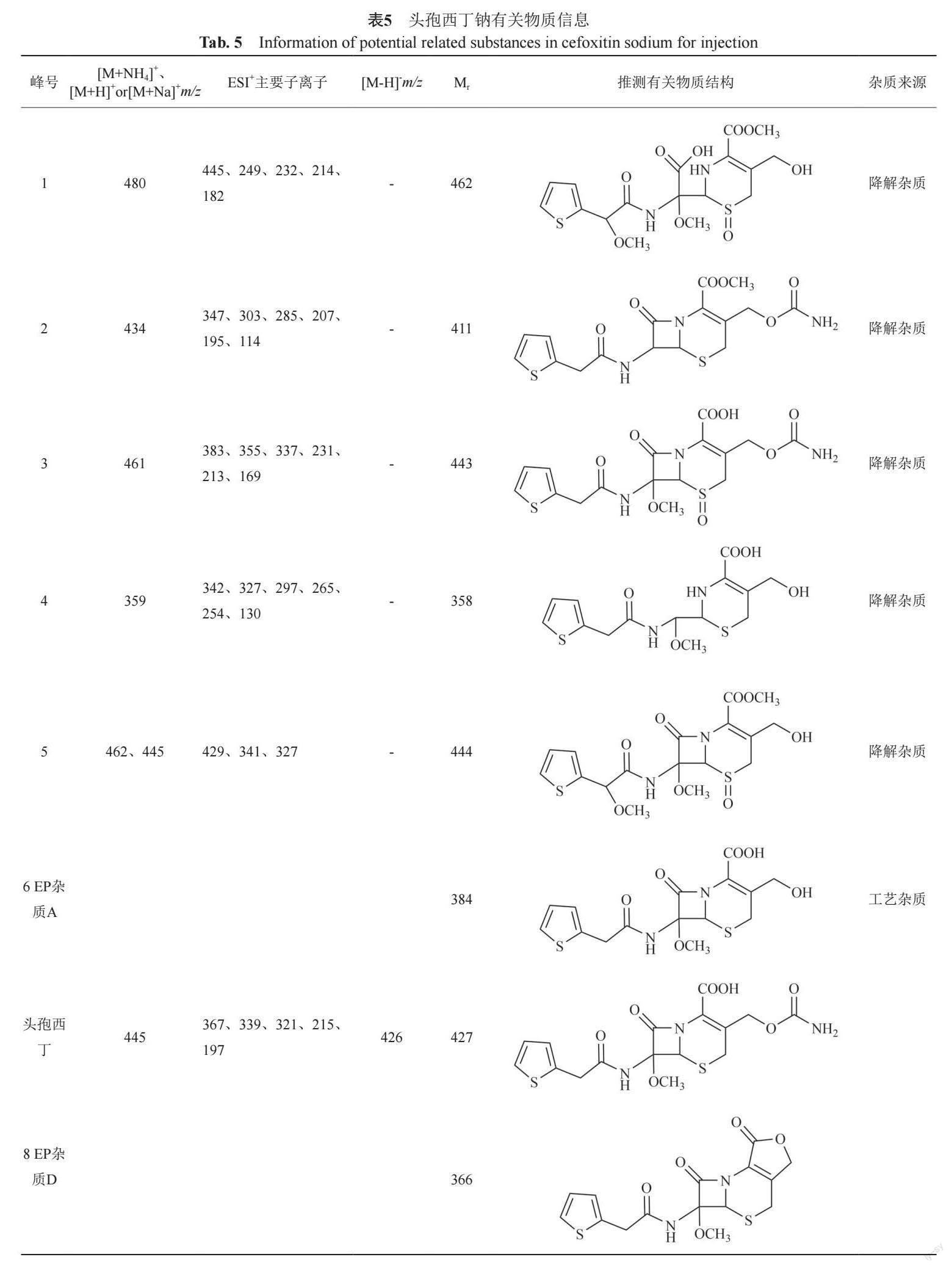
对强制降解溶液和样品中发现的色谱峰进行质谱分析,利用质荷比信息,并结合头孢西丁钠的生产工艺和已有文献,对样品中部分杂质的可能结构进行了推定。注射用头孢西丁钠中可能含有11个潜在杂质(表5),其中峰1、峰4和峰5的结构尚未见其他文献报道。
3 讨论
注射用头孢西丁有关物质分析中,无论采用何种药典标准方法,均有多对杂质的分离度未达到要求,本文经色谱柱筛选和CCD设计优化后,在60min内可使得各杂质峰的分离度达到最大值。与传统的试错法相比,采用计算机辅助设计优化,通过逐步缩小优化的区间,不仅可以最大程度减少实验次数,还可以考察不同因素的交互作用,提高预测准确度。
实验中发现,影响头孢西丁有关物质分析的主要影响因素为色谱柱参数H,色谱柱的疏水性越大,越易实现目标因子分离的要求。如在1号色谱柱(H值为0.623)中,目标因子R3/4和R8/9分别为1.513和0.219,而在6号色谱柱(H值为1.007)中,对应的分离度可以达到6.337和7.994;其次为流动相的pH、乙腈的比例、柱温和梯度时间。各因素对诸目标因子的影响不同,且存在交互作用,在部分区域甚至存在冲突。采用整体满意度函数,在确定的主要影响因素区间范围内,可以筛选到最优的色谱条件,使各目标因子同时满足要求。
制备混合系统适用性溶液时,分别考察了酸、碱、氧化、高温溶液和高温粉末五种强制降解条件。酸浓度增加至2 mol/L处理2 h时杂质峰无明显变化;高温粉末80℃放置13 d,仅峰6和峰8略微增加;0.2 mol/L碱溶液降解30 min时,峰2和峰11增加明显,其余杂质无明显变化,时间稍延长,主成分几乎全部降解;高温溶液80℃放置1 h,峰2、峰4、峰5和峰6均增加明显;3%氧化降解2 h,峰1、峰2和峰3均增加明显;基于上述试验结果,最终确定混合系统适用性溶液由溶液通过氧化和高温降解产生。
注射用头孢西丁钠在生产、运输和储藏过程中均会产生多种杂质。图1中,除峰6为工艺杂质外,其余均为降解杂质,峰7为氧化降解产生,其结构有待进一步确认。峰1、峰2和峰3均在氧化破坏后增加,峰2、峰4、峰5和峰6均在高温后有所增加。
4 结论
与常规方法不同,本文不再根据药典标准中规定的色谱柱类型(如EP中的苯基柱)去试错筛选色谱柱,而是从柱参数出发,考察其与目标因子的函数关系,确定某个参数或某几个参数的关键作用。并将计算机辅助设计与实际样品分离分析相结合,确定影响色谱分离的显著因素,考察各个因素的交互作用,成功地选择出头孢西丁钠有关物质分析的最优色谱条件,也为其他药物HPLC分析的优化提供了范例。本文对于完善注射用头孢西丁钠质量标准,考察不同企业的杂质谱差异,具有参考意义。
参 考 文 献
Lau W Y, Fan S T, Chu K W, et al. Cefoxitin versus gentamicin and metronidazole in prevention of post-appendicectomy sepsis: A randomized, prospective trial[J]. Antimicrob Chemother, 1986, 18(5): 613-619.
Sirinek K R, Levine B A. Antimicrobial management of surgically treated gangrenous orperforated appendicitis: comparison of cefoxitin and clindamycin-gentamicin[J]. Clin Ther, 1987, 9(4): 420-428.
Kim M H, Lee J M. Diagnosis and management of immediate hypersensitivity reactions to cephalosporins[J]. Allergy Asthma Immunol Res, 2014, 6(6): 485-495.
胡昌勤, 張夏. 化学药品杂质谱控制的现状与展望[J]. 药学学报, 2019, 54(12): 2214-2231.
赵山山. 注射用头孢西丁钠中有关物质的测定[J]. 河北化工, 2010, 33(7): 72-73.
朱庆珍, 庄凤侠. 头孢西丁钠有关物质的测定[J]. 光谱实验室, 2009, 26(6): 1449-1451.
李娜, 朱平, 王志敏. HPLC法测定注射用头孢西丁钠中有关物质[J]. 山西化工, 2008, 28(3): 29-31.
蒋秋玲, 石金芳, 狄斌, 等. LC-MS/MS法分析注射用头孢西丁钠中的有关物质[J]. 药物分析杂志, 2012, 32(9): 1597-1605.
肖慧, 洪建文, 彭洁, 等. 国产注射用头孢西丁钠质量评价[J]. 中国抗生素杂志, 2017, 42(6): 470-475.
薛晶, 朱克旭, 胡昌勤. 注射用头孢西丁钠的杂质谱比较[J]. 中国抗生素杂志, 2016, 41(8): 606-613.
王强, 李香荷, 韩彬, 等. 注射用头孢西丁钠的质量评价[J]. 中国抗生素杂志, 2022, 47(2): 134-139.
李进, 姚尚辰, 尹利辉, 等. 注射用头孢西丁钠的聚合物杂质分析[J]. 中国新药杂志, 2021, 30(11): 1038-1047.
国家药典委员会. 中华人民共和国药典. 二部[S]. 北京: 中国医药科技出版社, 2020: 304.
The United States Pharmacopieial Convention. The United States Pharmacopeia: General Chapters[S]. 43th edition. Rockville: The United States Pharmacopieial Convention, 2020: 858.
European Pharmacopoeia Commission. European Pharmacopoeia[S]. 10.0 edition. France. European Directorate for Quality Medicines, 2019: 2121-2125.
Neue U D. Stationary phase characterization and method development[J]. Sep Sci, 2007, 30(11): 1611-1627.
Snyder L R, Dolan J W, Carr P W. The hydrophobic-subtraction model of reversed-phase column selectivity[J]. Chromatogr A, 2004, 1060(1-2): 77-116.
