抗耐药结核新药贝达喹啉、德拉马尼和普托马尼的药动学特征及药物相互作用研究进展
2023-06-11丁杨明陆宇
丁杨明?陆宇
摘要:结核病(Tuberculosis, TB)是人类健康的主要威胁之一,多药组合的化学治疗是控制结核病的主要手段。近年贝达喹啉(Bedaquiline, BDQ)、德拉马尼(Delamanid, DLM)、普托马尼(Pretomanid, PMD)等抗耐药结核新药的出现,显著提高了耐药结核的治愈率。了解抗耐药结核新药的药动学特征及药物相互作用特点,合理设计药物组合及给药方案,可以减少药物不良反应发生,提高治疗成功率。本文对三种主要抗耐药结核新药的代谢动力学及药物相互作用研究进行综述,旨在为临床治疗提供依据和参考。
关键词:贝达喹啉;德拉马尼;普托马尼;药动学;药物相互作用
中图分类号:R978.3文献标志码:A
Abstract Tuberculosis (TB) is one of the most threats to human health, Chemotherapy with regimen of anti-tuberculosis drugs is the main means of controlling tuberculosis. The emergence of new anti-drug-resistant tuberculosis drugs such as bedaquiline, delamanid, and pretomanid has significantly increased the cure rate of drug-resistant tuberculosis. Research on the pharmacokinetics and drug-drug interactions of new anti-drug-resistant tuberculosis drugs, and designing drug and dosing regimens can reduce the incidence of adverse drug reactions and enhance the possibility of successful drug-resistant tuberculosis treatment. This article reviews the studies on the metabolism and drug interactions of three major new anti-drug-resistant tuberculosis drugs in recent years that provide a basis for clinical treatment and follow-up research.
Key words Bedaquiline; Delamanid; Pretomanid; Pharmacokinetics; Drug-drug interactions
結核病仍是人类健康的主要威胁之一,世界卫生组织(World Health Organization, WHO)估计,2019年全球新发结核病患者996万例,死亡结核病患者121万例,我国新发肺结核83.3万例,死亡3.1万例。其中,我国新发结核病7.1%为耐药或耐多药结核(multidrug-resistant tuberculosis, MDR-TB),目前估算我国有10~15万耐药结核病患者[1]。多药组合的化学治疗是控制结核病的主要手段,近年来,贝达喹啉(bedaquiline, BDQ)、德拉马尼(delamanid, DLM)、普托马尼(pretomanid, PMD)等新型抗耐药结核药物的相继上市,显著提升了耐药结核的治愈率,并缩短了治疗时间[2]。
药物相互作用(drug-drug interaction, DDI)是指一种药物的代谢及效应受到另一种药物,食物或者环境的影响而发生改变的现象。2020年9月,国家药监局颁布了我国首部《药物相互作用研究技术指导原则》[3],近年来新型抗结核药物的相互作用研究愈发受到重视。抗结核药物间的相互作用,MDR-TB与人免疫缺陷病毒(human immunodeficiency virus, HIV)共感染导致抗HIV药物与抗结核药物间的相互作用及患者合并病症治疗药物与抗结核药物间的相互作用,对新型抗结核药物临床应用形成了挑战。
随着BDQ、DLM和PMD等抗耐药结核新药的药动学及相互作用研究不断深入,本文将对三种新药的药动学特征及与抗结核药物和抗HIV药物间的药物相互作用进行综述,旨在为耐药结核的临床治疗提供依据及为药物组合的开发提供参考。
1 贝达喹啉(BDQ)
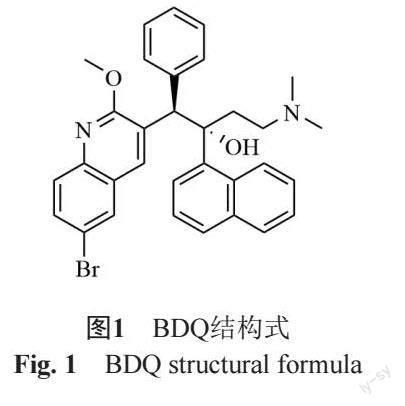
BDQ为二芳基喹啉类药物,结构式见图1。其与结核分枝杆菌(M. tuberculosis, MTB)的三磷酸腺苷(adenosine triphosphate, ATP)合成酶c亚基相结合,导致ATP合成受阻,从而阻止MTB中ATP能量供应,发挥抗菌作用。因其与传统抗结核药物作用机制不同,无交叉耐药性[4]。
1.1 BDQ的药动学特征
BDQ在治疗剂量下具有线性药动学特征,口服给药后,血药浓度达峰时间为5 h,食物可促进吸收,与标准餐同服时,BDQ的相对生物利用度将增加两倍[5],BDQ的绝对生物利用度在个体间差异较大,具体数值目前尚不明确[6]。BDQ的血浆蛋白结合率高达99.9%,表观分布容积约为164 L,体内血浆半衰期约为173 h,因组织中BDQ的缓慢释放,多剂量给药后,BDQ及其主要代谢物的终末半衰期长达5.5个月[7]。
BDQ在体内主要在肝脏经细胞色素P450超家族(cytochrome P450 proteins, CYP)中的CYP3A4途径代谢,部分经CYP2C8、CYP2C19代谢,生成多种代谢物M1~M8[8]。去甲基代谢物M2为其主要代谢物,其体内暴露量约为其母体化合物的23%~31%,M2暴露量与BDQ的主要不良反应QT间期延长的发生显著相关,且抗菌活性为原型的1/4~1/6之间[9]。BDQ对Ⅰ相代谢酶CYP酶系及Ⅱ相代谢酶UGT酶系皆无抑制作用[10-11]。
BDQ对OATP1B1、OATP1B3、BCRP、OAT1、OAT3等主要转运体均无抑制作用[12]。BDQ及其代谢物主要通过粪便排出,极少通过肾脏清除。
1.2 BDQ相互作用研究
1.2.1 BDQ与抗结核药物间相互作用
临床DDI研究显示,BDQ对多种抗耐药结核背景药物(异烟肼、吡嗪酰胺、乙胺丁醇、左氧氟沙星和环丝氨酸)的体内暴露量及代谢过程无显著影响,卡那霉素与BDQ联用时,卡那霉素的药-时曲线下面积(AUC)及最高血药浓度(Cmax)上升30%~50%[13]。BDQ为DDI受试药时,多种CYP酶抑制剂及诱导剂对BDQ及主要代谢物M2体内暴露量有显著影响。与强CYP3A4诱导剂利福平及利福喷汀联用时,BDQ的AUC及Cmax降低40%~60%。吡嗪酰胺及异烟肼对BDQ的体内暴露量无显著影响,但主要代谢物M2体内暴露量升高30%[13],抗耐药结核药物氯法齐明为CYP3A4的中到强抑制剂[14],群体药动学研究显示氯法齐明对BDQ及其主要代谢物M2的清除率和分布容积无显著影
响[15],但氯法齐明对BDQ及其代谢物暴露量的影响目前尚无研究报道。
1.2.2 BDQ与抗HIV药物间相互作用
抗HIV药物克力芝(洛匹那韦/利托那韦复方制剂)为目前主要抗耐药HIV药物,利托那韦通过抑制经CYP3A4途径代谢药物的清除,增强包括洛匹那韦在内的多种联用药物活性。利托那韦作为增效剂,已成为多个抗HIV及抗COVID-19方案中的重要组成部分[16]。
药动学研究显示,因利托那韦抑制BDQ主要代谢途径CYP3A4酶的活性,联用克力芝可增大BDQ暴露量30%以上[11]。临床DDI研究表明,联用多剂量克力芝后,患者体内BDQ暴露量上升一倍,M2暴露量无显著变化。利托那韦对BDQ及代谢物的Cmax无显著影响,但使其半衰期延长80%[17],群体药动学研究显示,BDQ与克力芝联用时,BDQ的清除率降低至单用时的25%左右,同时M2的清除率降低为59%[18]。
依法韦仑为抗艾滋一线药物,为CYP3A4诱导剂。研究显示,BDQ与依法韦仑联用时,BDQ的AUC降低了18%,而Cmax无显著变化,M2的AUC无显著变化,Cmax上升89%。患者群体药动学研究显示,随着BDQ与依法韦仑联用时间增长,BDQ的AUC可降低至单用时的50%以下[19],临床治疗中应对两药联用时的抗菌疗效进行关注。BDQ对多种抗HIV药物(洛匹那韦、利托那韦、尼韦西平、依法韦伦)代谢均无影响。抗艾滋药奈韦拉平对CYP3A4同时存在抑制和诱导的双重作用,BDQ与其联用时,清除率为单用时的82%,主要代謝物M2的清除率为单用时的119%,表明奈韦拉平与BDQ没有临床意义上的强相互作用[18-19]。
1.2.3 BDQ与其他药物间相互作用
葡萄柚汁中的呋喃香豆素对CYP3A4的抑制作用将显著增加患者BDQ的体内暴露[7]。除天然成分中广泛CYP酶抑制剂外,临床前研究显示,姜黄素及其类似物CC-I,6-丁香酚等天然产物单体将显著增加BDQ体内暴露,但具体机制尚需研究[20]。维拉帕米为质子泵抑制剂,可抑制MTB外排泵对药物的转运,为潜在的抗结核药物增效剂,其同时抑制CYP3A4的活性,研究显示,联用维拉帕米增加了BDQ的体内暴露,并显著增强了BDQ的抗结核活性[21]。
2 德拉马尼(DLM)
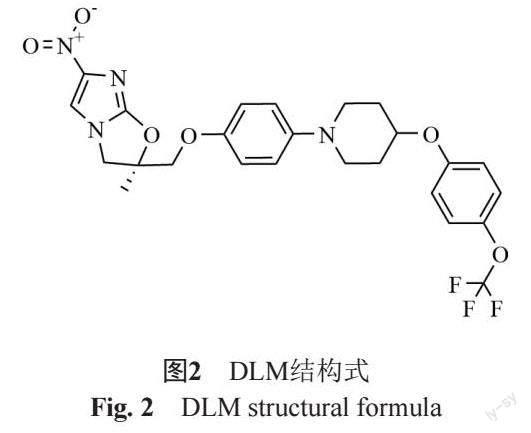
DLM为硝基咪唑类药物,结构式见图2。其在结核分枝杆菌内经F420代谢转化,生成其活性代谢产物发挥药效。除在分枝杆菌中代谢转化为活性代谢物外,DLM在体内发生广泛转化并生成多种代谢物 [22]。
2.1 DLM的药动学特征
DLM在治疗剂量下,呈线性药动学特征。当剂量高于200 mg时,呈非线性药动学特征。DLM的生物利用度为25%~47%,与食物同服可显著增加生物利用度,其血浆蛋白结合率高达99.5%以上[23]。DLM在体内主要经白蛋白代谢转化,部分于肝脏经CYP3A4途径代谢,较少部分经CYP1A1、CYP2D6 和 CYP2E1代谢,生成8种代谢物M1~M8[24]。DLM主要代谢物DM-6705(M1)的暴露量与DLM主要不良反应QT间期延长的发生显著相关[25],M1在体内经CYP途径代谢,进一步生成M2及M3。DLM的血浆半衰期在30~38 h之间,几乎不经尿液排泄途径消除[23]。
2.2 DLM的相互作用研究
DLM对CYP酶系无抑制或诱导作用[26],DLM非ABC转运蛋白、P-糖蛋白(P-gp)、有机阳离子转运多肽、有机阴离子转运多肽、乳腺癌耐药转运蛋白(BCRP)及溶质载体(SLC)转运蛋白在内的多种常见转运体的底物。DLM的主要代谢物M1为P-gp的底物,代谢物M1和M2在高浓度下抑制P-gp和BCRP介导的转运,但其抑制浓度远高于血浆中最大浓度,M3和 M4不影响任何转运蛋白的活动[27]。DLM对转运蛋白介导的吸收及外排无影响。
DLM与异烟肼,吡嗪酰胺,利福平三药联用时,DLM暴露量降低45%以上,乙胺丁醇在与DLM联用时,DLM的体内暴露无影响,乙胺丁醇体内暴露量上升25%[28]。
健康志愿者人群药动学研究显示,依法维伦等弱CYP诱导剂对DLM的体内暴露无影响。DLM与强P450酶抑制作用的利托那韦联用时体内暴露量上升25%[29]。
3 普托马尼(PMD)
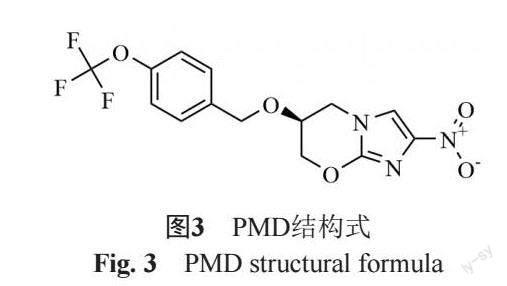
PMD同为硝基咪唑类药物,结构式见图3。其经非复制期MTB的F420代谢转化为活性代谢产物发挥药效。对复制期结核杆菌其主要作用机制是通过抑制分枝菌酸的形成及靶向MTB的磷酸戊糖途径[30]。
3.1 PMD的药动学特征
PMD在50~200 mg治疗剂量内呈线性药动学特征,餐后服用显著增加生物利用度[31]。血浆蛋白结合率约为86.4%,分布容积为130 L左右[32]。PMD主要以代谢物的形式经尿液及粪便进行排泄,仅1%以原形药物的形式经尿液的形式排出体外[33]。
3.2 PMD相互作用研究
PMD部分经CYP3A4途径代谢,生成多种代谢物,PMD无CYP抑制及诱导活性,其非OAT1、OAT3、OCT2、OAT1B1、OATP1B3、MATE1、MATE2-K、BCRP、P-gp等转运体的底物,同时对上述转运体无抑制作用[34]。
3.2.1 PMD与抗结核药物间相互作用
Ⅰ期临床DDI研究表明,联用利福平将使PMD的体内暴露量降低66% [35],患者群体药动学的研究显示,联用利福平或利福布汀后,PMD的清除率分别上升了80%、146%,同时PMD的暴露量分别下降44.4%、59.3%[36],研究显示利福布汀对PMD的体内暴露较利福平影响更明显。
在健康大鼠模型中,PMD、莫西沙星、吡嗪酰胺三药联用后,PMD与莫西沙星的体内暴露量显著上升,吡嗪酰胺的体内暴露量无变化[37],另一项大鼠模型研究显示,PMD与莫西沙星聯用时,PMD的体内暴露量显著上升,莫西沙星的体内暴露量不变[38]。目前临床研究尚无莫西沙星,吡嗪酰胺与PMD间DDI作用对其代谢动力学影响的报道。
3.2.2 PMD与抗HIV药物间相互作用
临床DDI研究表明,与多剂量依法韦仑联用时,PMD的体内暴露下降35%,联用利托那韦时,PMD的体内暴露下降17%。研究者认为,当联用利托那韦时,PMD的剂量无需调整[35]。健康大鼠模型中,联用达芦那韦将显著降低PMD的体内暴露,同时延长PMD的消除半衰期,然而其机制目前尚不明确,仍需临床DDI研究进行进一步验证[39]。
3.2.3 PMD与其他药物物间相互作用研究进展
临床研究显示,联用PMD对米达唑仑的体内暴露无影响,米达唑仑为CYP3A4探针底物,其体内暴露量反应联用药物对CYP3A4的抑制强度,研究结果显示,PMD及其代谢物对CYP3A4无抑制作用[40]。
BDQ、DLM、PMD 3种抗耐药结核新药均为强脂溶性药物,其在体内均呈现广泛分布、血浆蛋白结合率较高、消除半衰期长的代谢特征,三药在餐后服用可以提高其生物利用度。BDQ、DLM、PMD均经CYP途径代谢,联用CYP酶诱导剂利福霉素类药物或抗HIV药物依法韦仑将显著降低其体内暴露。CYP酶抑制剂或诱导剂与BDQ等药物间的DDI将增加抗结核治疗的安全性风险。当患者存在HIV与MDR-TB共感染并选择克力芝进行抗HIV治疗时,通过基于治疗药物监测结果,调整BDQ剂量,利托那韦作为增效剂,其可同时增强洛匹那韦与BDQ的药效,并降低因毒性代谢物M2带来的安全性风险。目前,抗耐药结核新药除与抗HIV药物及抗结核药物间相互作用研究外,尚无与其他药物间的相互作用研究。临床治疗中,除HIV与MDR-TB的共感染外,糖尿病,高血压等合并病症用药与抗耐药结核新药间的相互作用也引起了研究者们的关注[10]。对患者抗耐药结核新药的血药浓度及其代谢物浓度进行治疗药物监测,并及时调整给药剂量,可进一步提高治疗效果及患者对药物的耐受。
进一步开展抗耐药结核药物的相互作用研究具有重要意义。首先,与现有及潜在临床联用药物的DDI研究可提升治疗的安全性与有效性。其次,对新药代谢及DDI研究可为药物的开发及结构修饰的成药性研究提供参考。最后,从联用药物对抗耐药结核药物及其代谢物的体内暴露及抗结核活性的影响出发,可为开发新型药物组合及药物增效剂提供理论依据。
参 考 文献
World Health Organization (WHO).Global tuberculosis report 2020[R]. Geneva: World Health Organization, 2020.
WHO consolidated guidelines on drug-resistant tuberculosis treatment[M]. Geneva: World Health Organization, 2019.
药物相互作用研究技术指导原则(征求意见稿)[Z].国家药品监督管理局药品审评中心,2020 .
抗结核新药贝达喹啉临床应用专家共识(2020年更新版)[J]. 中华结核和呼吸杂志, 2021, 44(2): 81-87.
Matteelli A, Carvalho A C, Dooley K E, et al. TMC207: the first compound of a new class of potent anti-tuberculosis drugs[J]. Future Microbiol, 2010, 5(6): 849.
Svensson E M, du Bois J, Kitshoff R, et al. Relative bioavailability of bedaquiline tablets suspended in water: Implications for dosing in children[J]. Brit J Clin Pharm, 2018, 84(10): 2384.
US FDA. Briefing Package: NDA 204-384: Sirturo. [EB/OL]. (2014-08-16)[2014-10-04]. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/204384orig1s000sumr.pdf.
Liu K, Li F, Lu J, et al. Bedaquiline metabolism: Enzymes and novel metabolites[J]. Drug Metab Dispos, 2014, 42(5): 863.
Rouan M C, Lounis N, Gevers T, et al. Pharmacokinetics and pharmacodynamics of TMC207 and its N-desmethyl metabolite in a murine model of tuberculosis[J]. Antimicrob Agents Chemother, 2012, 56(3): 1444-1451.
Hu M, Zheng C, Gao F. Use of bedaquiline and delamanid in diabetes patients: Clinical and pharmacological considerations[J]. Drug Des Devel Ther, 2016, 10: 3983.
Cao L, Greenblatt D J, Kwara A. Inhibitory effects of selected antituberculosis drugs on common human hepatic cytochrome P450 and UDP-glucuronosyltransferase enzymes[J]. Drug Metab Dispos, 2017, 45(9): 1035-1043.
World Health Organization. The use of bedaquiline in the treatment of multidrug-resistant tuberculosis: interim policy guidance[M]. World Health Organization, 2013.
Van Heeswijk R P G, Dannemann B, Hoetelmans R M W. Bedaquiline: A review of human pharmacokinetics and drug-drug interactions[J]. J Antimicrob Chemother, 2014, 69(9): 2310-2318.
Sangana R, Gu H, Chun D Y, et al. Evaluation of clinical drug interaction potential of clofazimine using static and dynamic modeling approaches[J]. Drug Metab Dispos, 2018, 46(1): 26-32.
Maartens G, Brill M J E, Pandie M, et al. Pharmacokinetic interaction between bedaquiline and clofazimine in patients with drug-resistant tuberculosis[J]. Int J Tuberc Lung Dis, 2018, 22(1): 26-29.
Zhong H, Wang Y, Zhang Z L, et al. Efficacy and safety of current therapeutic options for COVID-19-lessons to be learnt from SARS and MERS epidemic: A systematic review and meta-analysis[J]. Pharmacol Res, 2020, 157: 104872.
Pandie M, Wiesner L, McIlleron H, et al. Drug–drug interactions between bedaquiline and the antiretrovirals lopinavir/ritonavir and nevirapine in HIV-infected patients with drug-resistant TB[J]. J Antimicrob Chemother, 71(4): 1037-1040.
Brill M J E, Svensson E M, Pandie M, et al. Confirming model-predicted pharmacokinetic interactions between bedaquiline and lopinavir/ritonavir or nevirapine in patients with HIV and drug-resistant tuberculosis[J]. Int J Antimicrob Agents, 2017, 49(2): 212-217.
Dooley K E, Park J G, Swindells S, et al. Safety, tolerability, and pharmacokinetic interactions of the antituberculous agent TMC207 (bedaquiline) with efavirenz in healthy volunteers: AIDS Clinical Trials Group Study A5267[J]. J Acquir Immune Defic Syndr, 2012, 59(5): 455.
Kotwal P, Dogra A, Sharma A, et al. Effect of natural phenolics on pharmacokinetic modulation of bedaquiline in rat to assess the likelihood of potential food–drug interaction[J]. J Agric Food Chem, 2020, 68(5): 1257-1265.
Xu J, Tasneen R, Peloquin C A, et al. Verapamil increases the bioavailability and efficacy of bedaquiline but not clofazimine in a murine model of tuberculosis[J]. Antimicrob Agents Chemother, 2018, 62(1): e01692-17.
Heidary M, Khoshnood S, Taki E, et al. Mechanism of action, resistance, synergism, and clinical implications of delamanid against multi drug-resistant Mycobacterium tuberculosis[J]. Front Microbiol, 2021: 2835.
Szumowski J D, Lynch J B. Profile of delamanid for the treatment of multidrug-resistant tuberculosis[J]. Drug Des Devel Ther, 2015, 9: 677.
Sasahara K, Shimokawa Y, Hirao Y, et al. Pharmacokinetics and metabolism of delamanid, a novel anti-tuberculosis drug, in animals and humans: importance of albumin metabolism in vivo[J]. Drug Metab Dispos, 2015, 43(8): 1267-1276.
Dooley K E, Rosenkranz S L, Conradie F, et al. QT effects of bedaquiline, delamanid, or both in patients with rifampicin-resistant tuberculosis: a phase 2, open-label, randomised, controlled trial[J]. Lancet Infect Dis, 2021.
Shimokawa Y, Sasahara K, Yoda N, et al. Delamanid does not inhibit or induce cytochrome p450 enzymes in vitro[J]. Biol Pharm Bull, 2014, 37(11): 1727-1735.
Sasabe H, Shimokawa Y, Shibata M, et al. Antitubercular agent delamanid and metabolites as substrates and inhibitors of ABC and solute carrier transporters[J]. Antimicrob Agents Chemother, 2016, 60(6): 3497-3508.
EMA. Deltyba (delamanid): authorisation details.. [EB/OL]. (2015-08-05)[2014-08-26] http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002552/human_med_001699.jsp&mid=WC0b01ac058001d124.
Mallikaarjun S, Wells C, Petersen C, et al. Delamanid coadministered with antiretroviral drugs or antituberculosis drugs shows no clinically relevant drug-drug interactions in healthy subjects[J]. Antimicrob Agents Chemother, 2016, 60(10): 5976-5985.
Stancil S L, Mirzayev F, Abdel-Rahman S M. Profiling pretomanid as a therapeutic option for TB infection: Evidence to date[J]. Drug Des Devel Ther, 2021, 15: 2815.
Winter H, Ginsberg A, Egizi E, et al. Effect of a high-calorie, high-fat meal on the bioavailability and pharmacokinetics of PA-824 in healthy adult subjects[J]. Antimicrob Agents Chemother, 2013, 57(11): 5516-5520.
Lyons M A. Modeling and simulation of pretomanid pharmacokinetics in pulmonary tuberculosis patients[J]. Antimicrob Agents Chemother, 2018, 62(7): e02359-17.
Thakare R, Dasgupta A, Chopra S. Pretomanid for the treatment of pulmonary tuberculosis[J]. Drugs Today (Barc), 2020, 56(10): 655-668.
US FDA. Pretomanid FDA highlights of prescribing information [EB/OL]. (2019-10-12)[2019-10-13] https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212862Orig1s000Lbl.pdf.
Dooley K E, Luetkemeyer A F, Park J G, et al. Phase I safety, pharmacokinetics, and pharmacogenetics study of the antituberculosis drug PA-824 with concomitant lopinavir-ritonavir, efavirenz, or rifampin[J]. Antimicrob Agents Chemother, 2014, 58(9): 5245-5252.
Ignatius E H, Abdelwahab M T, Hendricks B, et al. Pretomanid pharmacokinetics in the presence of rifamycins: Interim results from a randomized trial among patients with tuberculosis[J]. Antimicrob Agents Chemother, 2021, 65(2): e01196-20.
Wang L, Xu Y, Liang L, et al. LC–MS/MS method for the simultaneous determination of PA-824, moxifloxacin and pyrazinamide in rat plasma and its application to pharmacokinetic study[J]. J Pharm Biomed Anal, 2014, 97: 1-8.
Jing J, Jia Y, Feng T, et al. Study on the drug–drug interaction of antituberculosis drugs—PA-824 and moxifloxacin based on pharmacokinetics in rats by LC–MS/MS[J]. Acta Chromatogr, 2019, 31(3): 206-210.
Wang L, Zhao J, Zhang R, et al. Drug-drug interactions between PA-824 and darunavir based on pharmacokinetics in rats by LC–MS-MS[J]. J Chromatogr Sci, 2018, 56(4): 327-335.
Winter H, Egizi E, Erondu N, et al. Evaluation of pharmacokinetic interaction between PA-824 and midazolam in healthy adult subjects[J]. Antimicrob Agents Chemother, 2013, 57(8): 3699-3703.
