经典名方温经汤基准样品HPLC-Q-TOF/MS分析与指纹图谱研究
2023-06-10汤志锋张云羽石德志高武锋龚小文嵇晶黄仕文程建明
汤志锋,张云羽,石德志,高武锋,龚小文,嵇晶,黄仕文,程建明
(1.南京中医药大学药学院,江苏 南京 210023;2.江苏省经典名方工程研究中心,江苏 南京 210023;3.南京中医药大学中医学院·中西医结合学院,江苏 南京 210023)
经典名方温经汤出自《妇人大全良方》,该方由酒当归、川芎、芍药、桂心、牡丹皮、醋莪术、人参、炒甘草、酒牛膝9味药组成,为妇科调经的常用方,主要用于冲任虚寒而有瘀滞的月经不调[1]、痛经[2-3]、崩漏、不孕[4]等。《古代经典名方关键信息表(7首方剂)》[5](以下简称关键信息表)对其用法用量有明确规定“粉碎成粗粒,每服20 g,加水450 mL,煎至240 mL,去滓温服”。本团队在关键信息表颁布后,完成了资源考察、产地优选、药材和饮片的质量研究以及基准样品的制备工艺研究,并在此基础上对温经汤基准样品进行质量研究。
目前温经汤的研究主要集中在临床应用方面,化学成分分析与指纹图谱的相关研究较少,尚未有关于温经汤基准样品化学成分分析与指纹图谱同时测定的相关报道。本方成分复杂,仅对部分指标性成分进行含量测定,难以代表全方,指纹图谱技术的运用能较为全面地表征复方复杂的化学成分信息[6-9],国家药监局药审中心发布的第36号文件[10]明确指出“指纹/图谱一般以相似度或特征峰相对保留时间、相对峰面积等为检测指标,主要成分在指纹/特征图谱中应尽可能得到指认,必要时应建立多张指纹/特征图谱”。目前温经汤的图谱研究尚未指认全方所用药味,本研究通过HPLC-Q-TOF/MS技术分析基准样品的化学成分组成,并采用HPLC-UV和HPLC-ELSD法建立温经汤基准样品的指纹图谱,首次归属了全方所有药味,为古代经典名方温经汤基准样品的质量控制和新药研发打下基础。
1 材料
1.1 仪器与试药
Waters e2695高效液相色谱仪(美国Waters公司),Waters 2998 PDA检测器(美国Waters公司),Triple TOF 5600型质谱仪(美国SCIEX公司),TD6002C型电子天平(天津天马衡基仪器有限公司),AUW120D型电子分析天平(日本岛津公司),KH-300E型超声波清洗器(昆山禾创超声仪器有限公司),FTS-10A液体加热器(潮州市一壶百饮电器实业有限公司),FW100型高速粉碎机(天津市泰斯特仪器有限公司)。
甘草苷、甘草酸铵、芍药苷、桂皮醛、丹皮酚、阿魏酸、没食子酸、β-蜕皮甾酮、人参皂苷Rg1、人参皂苷Rb1对照品(中国食品药品检定研究院,批号:111610-201607、110731-201720、110736-201035、110710-201821、110708-201407、110773-201313、110831-201906、111638-201706、110703-202034、110704-202129,纯度:93.1%、97.7%、97.7%、99.4%、99.9%、99.6%、91.5%、95.0%、94.0%、94.3%),人参皂苷Re、藁本内酯对照品(上海源叶生物科技有限公司,批号:B04D9S76499、R21J10F91128,纯度均≥98%)。
甲醇、乙腈(美国Tedia公司,色谱纯),超纯水(Unique-R10多功能超纯水系统制备),甲酸(上海阿拉丁生化科技股份有限公司,色谱纯)。
1.2 药材
温经汤所用饮片1批购自安徽省万生中药饮片有限公司,5批由实验室炮制而成,10批组方对应饮片批次见表1,实验室所用药材经南京中医药大学严辉教授鉴定,按照2020版《中国药典》方法检测,所用批次饮片质量均符合药典规定。
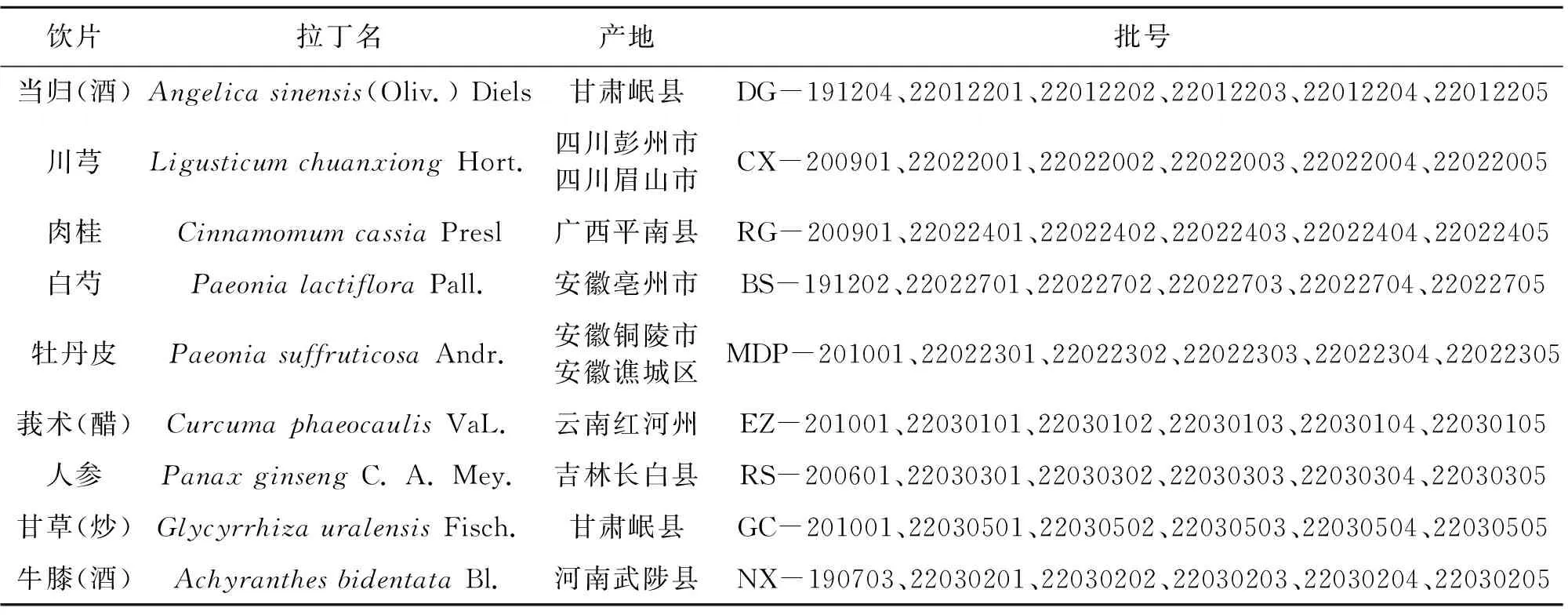
表1 温经汤基准样品饮片来源及批号信息Table 1 The origin and batch number of herbal pieces from WJT substance benchmarks
2 方法与结果
2.1 HPLC-Q-TOF/MS成分分析
2.1.1 色谱条件 Agilent 5 TC-C18(2)色谱柱(4.6 mm×250 mm,5 μm);流动相A为乙腈,B为0.1%甲酸水溶液,梯度洗脱(0~56.0 min,3.0%~61.2%A;56.0~56.1 min,61.2%~80%A;56.1~57.0 min,80%~90%A;57.0~65.0 min,90%A);检测波长254 nm;进样量为10 μL;流速0.8 mL·min-1;柱温30 ℃。
2.1.2 质谱条件 电喷雾离子源(ESI),采用正负离子扫描检测,质量扫描范围m/z50~1 500;喷雾电压45 kV;雾化气压力60 psi;辅助气压力60 psi;气帘气压力40 psi;离子源温度600 ℃;锥孔电压为100 V;碰撞室射出电压40 eV。
2.1.3 供试品溶液的制备 按关键信息表中温经汤的用法用量,称取每服处方量(各单味药批次随机组合成10批,编号为S1~S10),粉碎成最粗粉,加水450 mL(22.5倍量),浸泡30 min后武火至沸,转至文火,煎煮30 min(至240 mL),100目双层纱布趁热过滤,取滤液1 mL,加2 mL甲醇,混匀,1 000 r·min-1离心,取上清液过0.45 μm微孔滤膜,即得。
2.1.4 温经汤基准样品化学成分分析 使用上述色谱、质谱条件对供试品溶液进行正、负离子全扫描,获得正、负离子模式下的总离子流图(Total ion chromatogram,TIC图),见图1。将数据导入PeakView软件,从温经汤基准样品中共鉴别了122个化合物[11-16],具体见表2。
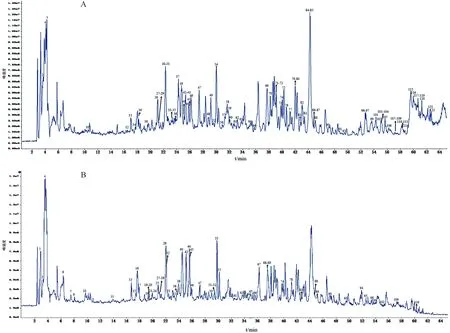
图1 温经汤正(A)、负(B)离子模式下的总离子流图Fig.1 Total ion chromatogram of WJT in positive (A) and negative (B) ion modes
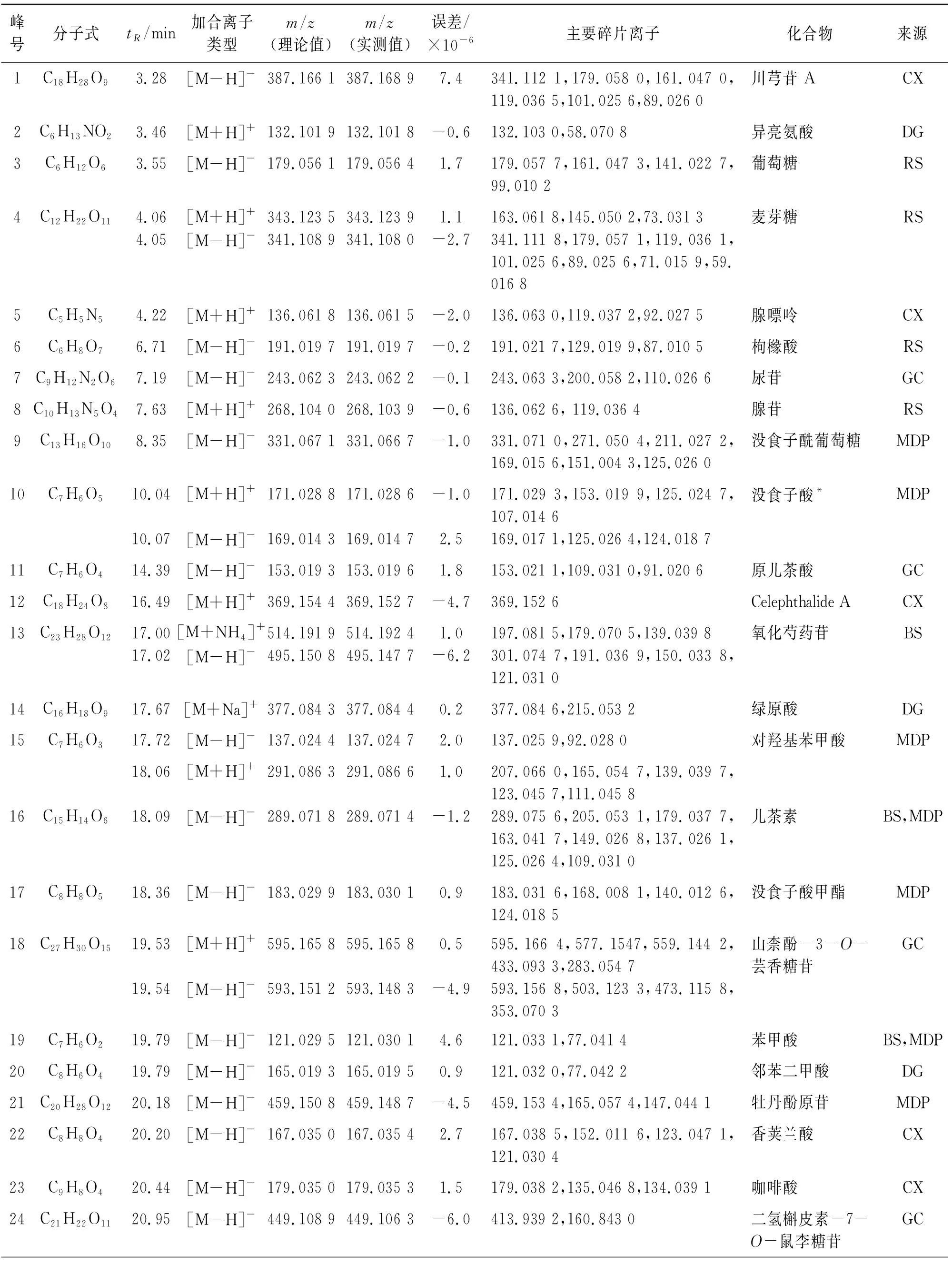
表2 温经汤基准样品HPLC-Q-TOF-MS分析Table 2 HPLC-Q-TOF-MS analysis results of WJT substance benchmarks
通过HPLC-Q-TOF/MS分析鉴别出122个化合物,包括有机酸、黄酮类、苷类、萜类、甾体、鞣质类等,对其药味来源进行归属,其中45个源自甘草,6个源自白芍,15个源自牡丹皮,13个源自川芎,10个源自当归,11个源自人参,1个源自牛膝,5个源自肉桂,9个源自莪术,化合物16、19、27、44、45为白芍、牡丹皮共有成分,化合物46和114为川芎、当归共有成分。
2.2 温经汤指纹图谱的建立
2.2.1 色谱条件 同“2.1.1”项下色谱条件。
2.2.2 供试品溶液的制备 同“2.1.3”项下供试品溶液的制备。
2.2.3 对照品溶液的制备 取没食子酸、芍药苷、β-蜕皮甾酮、甘草苷、阿魏酸、桂皮醛、丹皮酚、甘草酸、藁本内酯对照品适量,精密称定,加甲醇制成质量浓度分别为123.53、7.05、4.19、10.48、13.75、17.19、3.73、71.21、10.14 μg·mL-1的混合对照品溶液。
2.2.4 各单味药及阴性供试品的制备 分别称取处方中每服剂量所需相应饮片的量(S1中各批次单味药),按照“2.1.2”项下方法制备,得单味药供试品溶液。
分别称取处方中每服剂量所需相应的缺当归、缺肉桂、缺白芍、缺牡丹皮、缺牛膝、缺川芎、缺甘草、缺莪术、缺人参9个缺单味药和缺当归、肉桂,缺白芍、牡丹皮2个双缺药(S1中各批次单味药),按照“2.1.2”项下方法制备,得阴性对照供试品溶液。
2.2.5 方法学考察
2.2.5.1 精密度试验 取“2.1.3”项下制备供试品溶液,色谱条件同“2.1.1”,连续进样6针,以阿魏酸色谱峰为参照峰(S),计算各共有峰与S峰的相对保留时间和相对峰面积RSD值,各共有峰相对保留时间RSD小于1%,相对峰面积RSD小于5%,表明仪器精密度良好。
2.2.5.2 重复性试验 取每服饮片量,按照“2.1.3”项下制备得6份供试品溶液,按照“2.1.1”项下色谱条件进样分析,以阿魏酸色谱峰为参照峰(S),计算各共有峰与S峰的相对保留时间和相对峰面积RSD值,各共有峰相对保留时间RSD小于1%,相对峰面积RSD小于5%,表明重复性良好。
2.2.5.3 稳定性考察 取同一批供试品溶液,同“2.1.1”项下色谱条件分别于0、4、8、12、16、24 h进样测定,以阿魏酸色谱峰为参照峰(S),计算各共有峰与S峰的相对保留时间和相对峰面积RSD值,各共有峰相对保留时间RSD小于1%,相对峰面积RSD小于5%,表明样品在24 h内稳定。
2.2.6 温经汤基准样品指纹图谱相似度评价 按照“2.1.1”项的指纹图谱条件,按照“2.1.3”项的方法制备S1~S10供试品溶液,并进行HPLC-UV测定,得到10批温经汤HPLC-UV指纹图谱。将10批温经汤指纹图谱依次导入《中药色谱指纹图谱相似度评价系统(2012版)》软件,以S5为参照图谱,采用中位数法,时间窗宽度为0.1,进行多点校正,标注19个共有峰,并进行Mark峰匹配,生成对照图谱并计算相似度,10批温经汤基准样品指纹图谱的相似度均>0.910,具体数据见表3。温经汤对照指纹图谱(R)和10批温经汤叠加图谱(S1~S10)见图2。
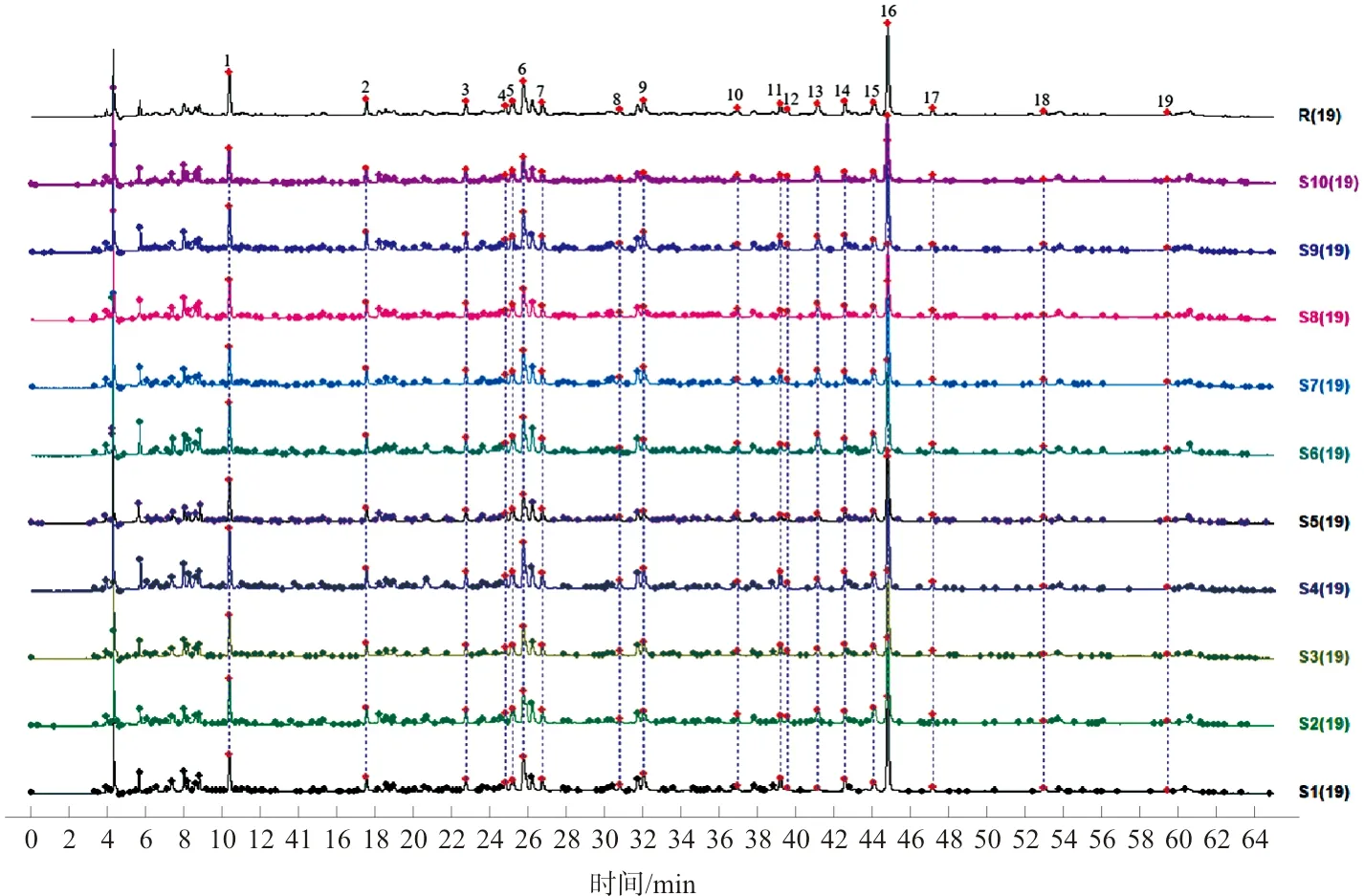
图2 温经汤对照指纹图谱(R)及10批温经汤叠加图谱(S1~S10)Fig.2 Reference fingerprint (R) and fingerprint of 10 batches of WJT (S1-S10)
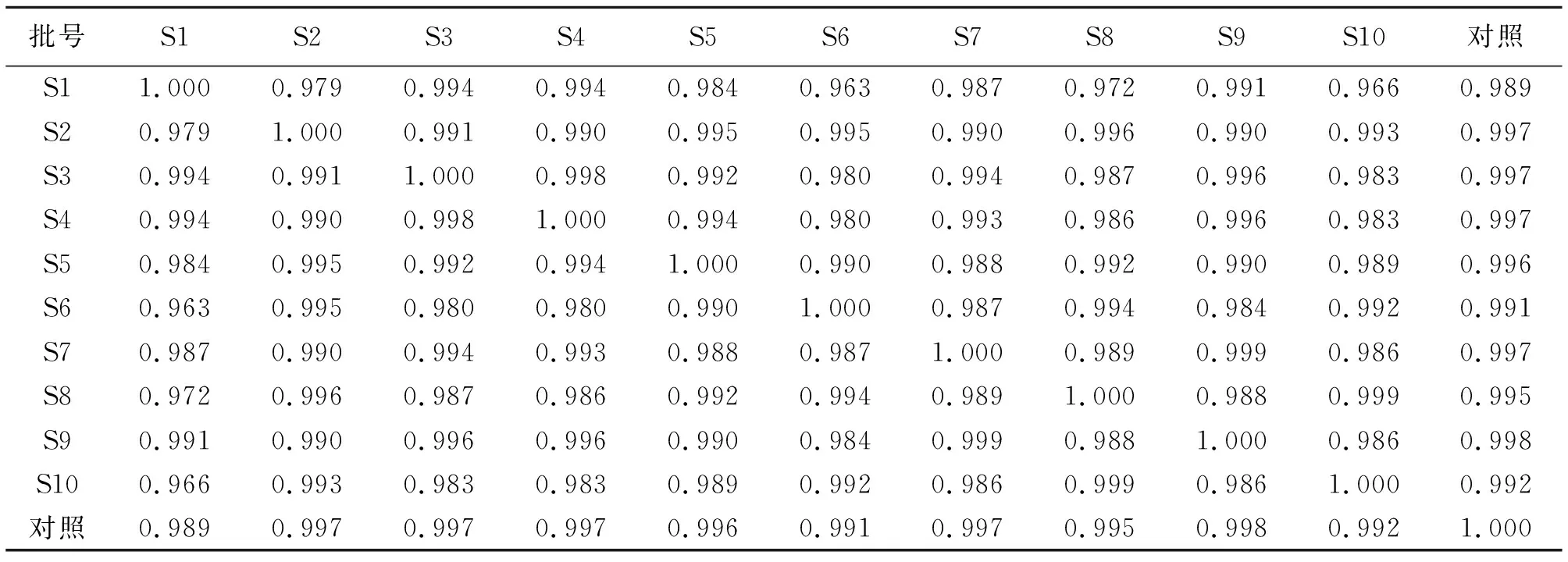
表3 10批温经汤基准样品HPLC-UV指纹图谱相似度Table 3 Similarity of fingerprint of 10 batches of WJT substance benchmarks
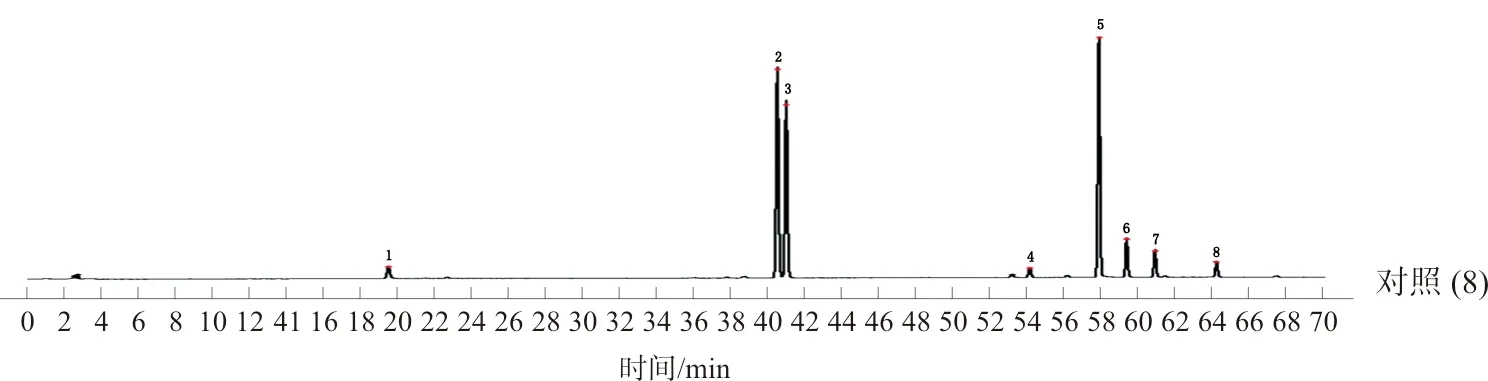
2.2.7 温经汤指纹图谱峰指认及峰归属 将温经汤指纹图谱与混合对照品溶液、单味药材供试品溶液及各药材阴性对照供试品溶液进行比对,指认出其中9个色谱峰(图3),并对19个共有峰进行归属(图4),其中10个峰来源于甘草(5、6、8、9、10、11、12、14、16、17),4个峰来源于牡丹皮(1、2、3、15),1个峰来源于白芍(3),1个峰来源于牛膝(4),2个峰来源于当归(7、19),2个峰来源于川芎(7、19),1个峰来源于肉桂(13),1个峰来源于莪术(18)。峰3是白芍和牡丹皮的共有成分,峰7和峰19是川芎和当归的共有成分,除人参药味尚无共有峰归属,其余药味均有归属。
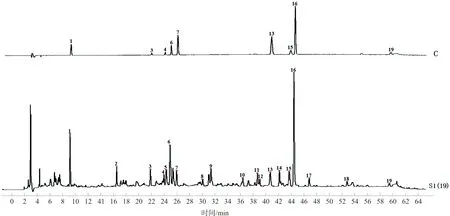
注:1.没食子酸;3.芍药苷;4.β-蜕皮甾酮;6.甘草苷;7.阿魏酸;13.桂皮醛;15.丹皮酚;16.甘草酸;19.藁本内酯图3 HPLC-UV混合对照品图(C)及温经汤对照指纹图谱(S1)对比图Fig.3 Comparison diagram of HPLC-UV mixed reference (C) and WJT reference fingerprint (S1)
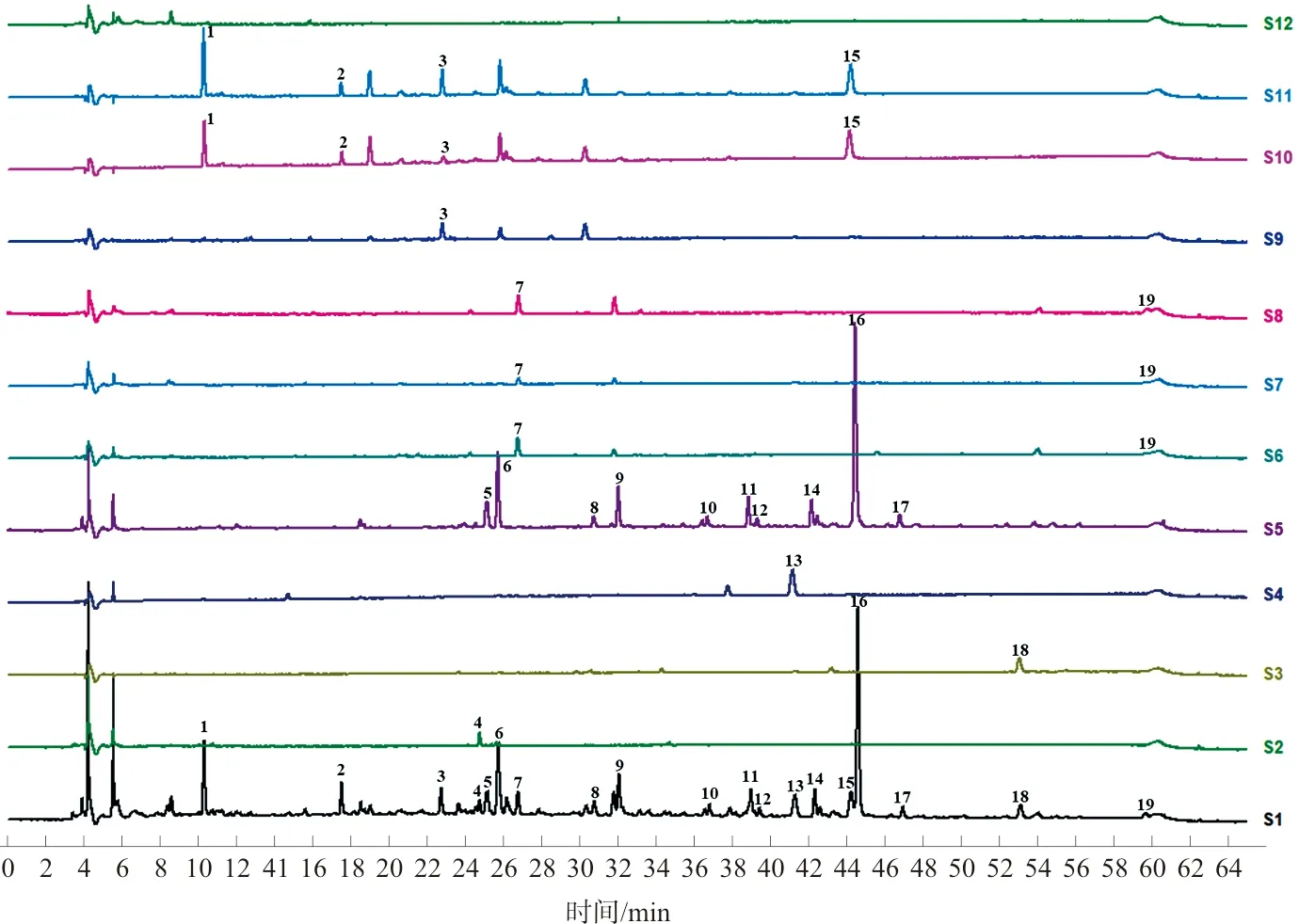
注:S1.全方;S2.牛膝;S3.莪术;S4.肉桂;S5.甘草;S6.川芎;S7.当归;S8.川芎和当归;S9.白芍;S10.牡丹皮;S11.白芍和牡丹皮;S12.人参图4 单味药材共有峰归属Fig.4 Common peak attribution of single medicine materials
2.2.8 温经汤参照峰的选择及相对保留时间的计算 综合10批温经汤基准样品保留时间结果,详见表4 ,确定了温经汤基准样品的鉴别标准,建议煎液指纹图谱中应有19个共有峰,并以保留时间及峰面积相对适中且相对稳定的阿魏酸(7号峰)作为参照峰(S峰),计算各共有峰与S峰的相对保留时间,其相对保留时间偏差应在规定值的±10%之内。规定值为:0.387(峰1)、0.655(峰2)、0.849(峰3)、0.927(峰4)、0.940(峰5)、0.962(峰6)、1.000(峰7)、1.150(峰8)、1.198(峰9)、1.381(峰10)、1.465(峰11)、1.478(峰12)、1.538(峰13)、1.591(峰14)、1.648(峰15)、1.675(峰16)、1.762(峰17)、1.980(峰18)、2.223(峰19)。本方挥发性成分较多,不同批次和来源饮片之间存在较大差异,相对峰面积RSD值相对偏高,除峰13(桂皮醛)、峰18(莪术)两峰相对峰面积>30%外,其他峰的相对峰面积均<30%。确定药材产地、批次以及具体炮制方法后,可全面把控全方的整体质量,缩小批次间差异。
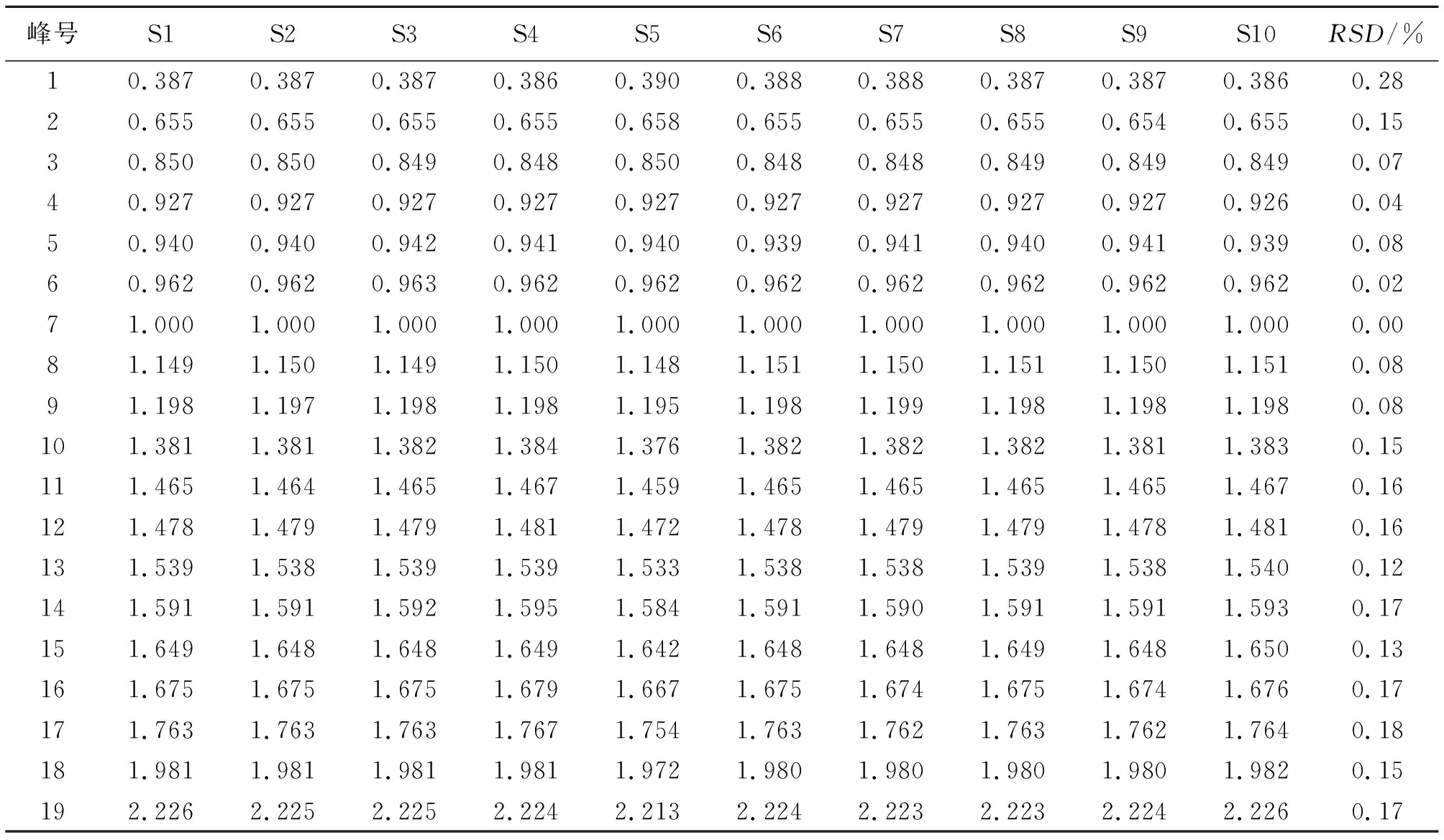
表4 10批温经汤基准样品共有峰相对保留时间Table 4 Relative shift of retention time of characteristic peaks in transfer among 10 batches of WJT
2.3 皂苷类成分的指纹图谱研究
在HPLC-UV法建立的温经汤基准样品指纹图谱中,因人参皂苷类成分具有末端吸收,与其他成分吸收波长有较大差异,尚未有人参共有峰的归属,为全面表征基准样品的整体特征,进一步对基准样品进行萃取纯化,通过HPLC-ELSD法,建立了1张能够表征人参的皂苷类成分指纹图谱[17-20],弥补HPLC-UV图谱中无法体现全方整体药味的不足。
2.3.1 色谱条件 Agilent 5 TC-C18(2)色谱柱(4.6 mm×250 mm,5 μm);流动相A为乙腈,B为水,梯度洗脱(0~30.0 min,15.0%~23.0%A;30.0~60.0 min,23.0%~40.0%A;60.0~70.0 min,40%A);漂移管温度90 ℃;载气体积流量1.6 L·min-1;进样量10 μL;流速1.0 mL·min-1;柱温25 ℃。
2.3.2 供试品溶液的制备 按“2.1.3”项下,称取温经汤每服处方量(S1~S10),粉碎,制备10批煎液基准样品,冷冻干燥,得基准样品冻干粉。精密称定冻干粉各5 g,精密加水50 mL,密塞,称定质量,超声30 min,冷却至室温,补足失质量,过滤。滤液用水饱和正丁醇萃取3次,每次50 mL,合并正丁醇萃取液,用2%NaOH洗2次,每次50 mL,再用蒸馏水50 mL洗2次,弃去水液,正丁醇层置水浴锅上蒸干,残渣加甲醇溶解并转移至10 mL量瓶中,加甲醇稀释至刻度,摇匀,过0.45 μm微孔滤膜,即得。
2.3.3 对照品溶液的制备 取人参皂苷Rg1、人参皂苷Re、人参皂苷Rb1对照品适量,精密称定,加甲醇制成质量浓度分别为1.012、0.258、1.035 mg·mL-1的对照品溶液。
2.3.4 方法学考察
2.3.4.1 精密度试验 取“2.3.2”项下制备供试品溶液,色谱条件同“2.3.1”,连续进样6针,以人参皂苷Rg1色谱峰为参照峰(S),计算各特征峰与S峰的相对保留时间和相对峰面积RSD值,各共有峰相对保留时间RSD小于1%,相对峰面积RSD小于5%,表明仪器精密度良好。
2.3.4.2 重复性试验 取每服饮片量,按照“2.3.2”项下制备得6份供试品溶液,按照“2.3.1”项下色谱条件进样分析,以人参皂苷Rg1色谱峰为参照峰(S),计算各特征峰与S峰的相对保留时间和相对峰面积RSD值,各共有峰相对保留时间RSD<1%,相对峰面积RSD<5%,表明重复性良好。
2.3.4.3 稳定性考察 取同一批供试品溶液,同“2.3.1”项下色谱条件分别于0、4、8、12、16、24 h进样测定,以人参皂苷Rg1色谱峰为参照峰(S),计算各共有峰与S峰的相对保留时间和相对峰面积RSD值,各特征峰相对保留时间RSD<1%,相对峰面积RSD<5%,表明样品在24 h内稳定。
2.3.5 皂苷类成分指纹图谱的建立 按照“2.3.2”项的方法制备S1~S10供试品溶液,按照“2.3.1”项下色谱条件,进行HPLC-ELSD测定,得到10批温经汤HPLC-ELSD指纹图谱。将10批温经汤基准样品HPLC-ELSD指纹图谱依次导入《中药色谱指纹图谱相似度评价系统(2012版)》软件,以S5为参照图谱,采用中位数法,时间窗宽度为0.1,进行多点校正,标注8个共有峰,并进行Mark峰匹配,生成对照图谱并计算相似度,10批HPLC-ELSD图谱的相似度均>0.950,对照指纹图谱(R)、10批温经汤HPLC-ELSD叠加图谱(S1~S10)及对照品图谱见图5~6。
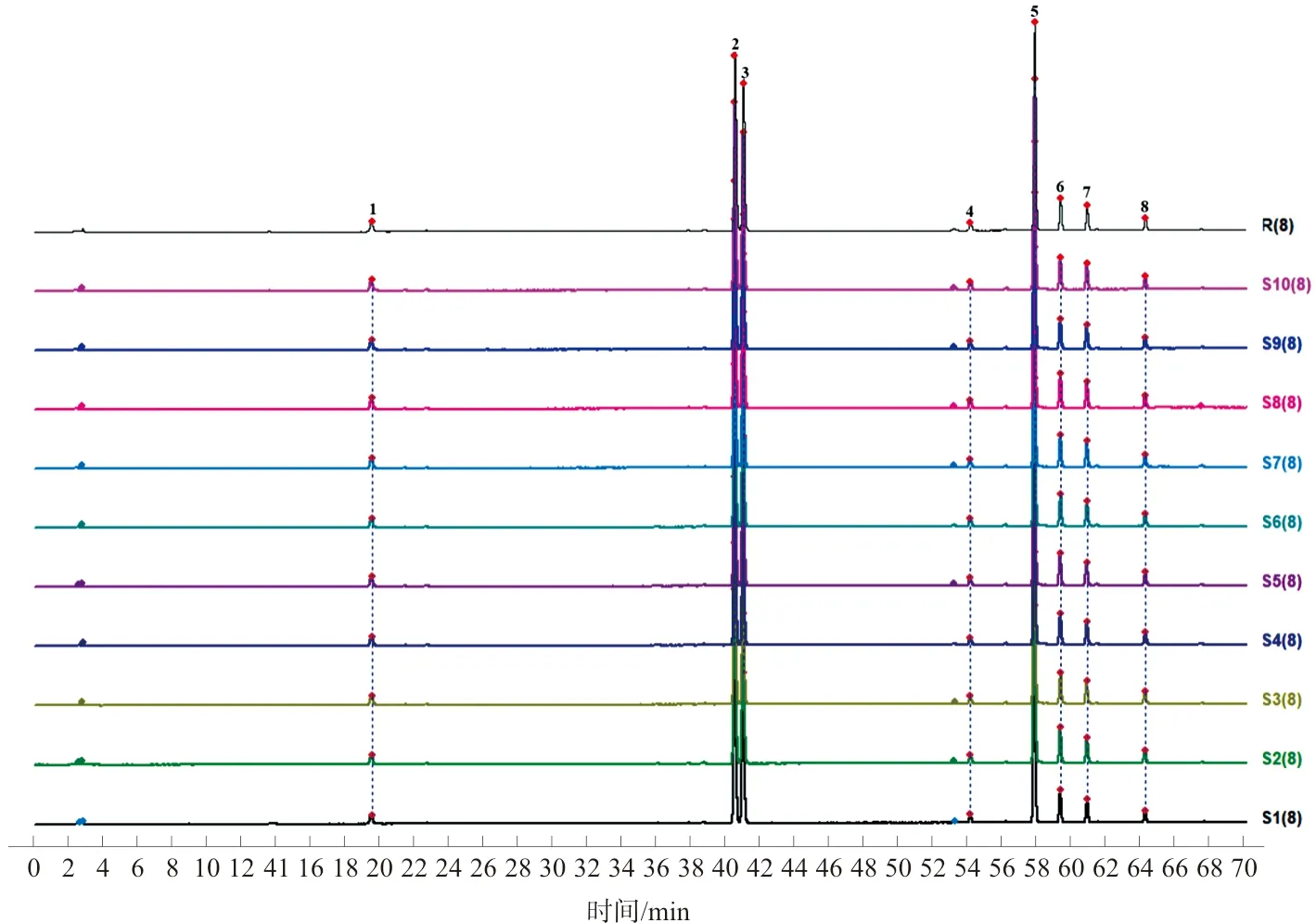
图5 温经汤对照指纹图谱(R)及10批温经汤ELSD叠加图谱(S1~S10)Fig.5 HPLC reference fingerprint (R) and ELSD fingerprint of 10 batches of WJT (S1-S10)
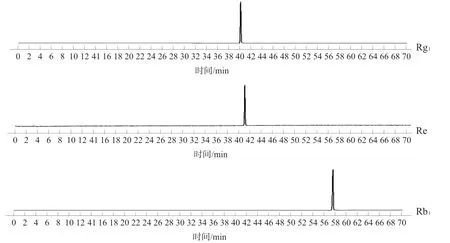
注:2.人参皂苷Rg1;3.人参皂苷Re;5.人参皂苷Rb1图6 HPLC-ELSD对照品(Rg1、Re、Rb1)图及温经汤对照指纹图谱对比图Fig.6 Comparison diagram of HPLC-ELSD of reference (Rg1, Re, Rb1) and WJT reference fingerprint
2.3.6 温经汤参照峰的选择及相对保留时间的计算 综合10批温经汤基准样品保留时间结果,确定了温经汤基准样品的鉴别标准,建议煎液指纹图谱中应有8个共有峰,并以保留时间及峰面积相对适中且相对稳定的人参皂苷Rg1(2号峰)作为参照峰(S峰),计算各共有峰与S峰的相对保留时间,结果10批温经汤8个共有峰相对保留时间RSD值<5%,相对峰面积RSD值均<10%。
3 讨论
本研究对建立的HPLC-UV指纹图谱色谱条件进行优化,单因素考察了不同柱温(25、30、35 ℃),不同流速(0.6、0.8、1.0 mL·min-1),不同进样量(10、20、30、40、50 μL),不同流动相(乙腈-甲酸、乙腈-醋酸、乙腈-磷酸),不同色谱柱[Agilent 5 TC-C18(2)、Agilent SB-C18、Kromasil 100-5-C18、Hedera ODS-2]对洗脱效果的影响,发现不同温度、不同进样量对图谱无较大影响,流速为0.6 mL·min-1,色谱峰较宽,随着流速的增大,各共有峰保留时间前移,流速为1.0 mL·min-1,部分色谱峰出现交叉,分离度较差。不同色谱柱对图谱的出峰数目以及分离度有较大影响,本研究所用Agilent 5 TC-C18(2)色谱柱出峰数目多,分离度好;Agilent SB-C18色谱柱有较好的分离度,但出峰数目较少;Kromasil 100-5-C18色谱柱,出峰数目少,且部分峰出现拖尾;Hedera ODS-2色谱柱中峰19(藁本内酯)分离度较其他色谱柱好,但甘草的相关共有峰出现交叉。根据出峰数目及分离效果,色谱最佳条件:色谱柱为Agilent 5 TC-C18(2),乙腈-甲酸为流动相,进行梯度洗脱,柱温30 ℃,流速0.8 mL·min-1,进样量10 μL。
通过单味药和缺阴性药供试品与复方供试品成分比对,将HPLC-UV指纹图谱中19个共有峰进行归属,共归属了除人参外的8味药材。因人参皂苷类成分与其它成分吸收波长存在较大差异,无法在HPLC-UV指纹图谱得到归属,针对人参皂苷类成分的末端吸收现象,对基准样品进行萃取纯化,建立HPLC-ELSD指纹图谱,通过对照品指认出人参的主要成分人参皂苷Rg1、人参皂苷Re和人参皂苷Rb1。当前有关经典名方温经汤基准样品的相关研究[7-8,21-22]中,尚未有通过指纹图谱指认出全方所有药味的明确报道。本研究建立的HPLC-UV和HPLC-ELSD指纹图谱,首次归属了全方所有药味,弥补了当前研究的空缺,为后续温经汤基准样品、制剂的质量控制及量值传递研究提供参考。
