全缘千里光碱脂质体及其凝胶剂的皮肤渗透研究与机制探讨
2023-06-08卢玉洁周珊珊单钰君徐莹莹程碧欣朱志红郑杭生
卢玉洁,周珊珊,单钰君,徐莹莹,程碧欣,朱志红,郑杭生
浙江中医药大学药学院,浙江 杭州 310053
全缘千里光碱(integerrimine,INT)是从菊科(Compositae)千里光属SenecioL.的多年生草本植物峨嵋千里光SeneciofaberiHemsl.中提取的一种双稠吡咯环生物碱,能干扰细胞的有丝分裂而具有抗肿瘤作用,但全身用药后,对肝脏产生蓄积性的、不可逆的强烈毒性作用[1]。除了在20 世纪70 年代国内临床上小规模地通过灌注治疗膀胱癌[2]外,基本未在临床推广使用。徐月红等[2]与成秉辰[3]的研究证明INT 对皮肤黑色素瘤有抑制作用,徐月红等[4]首先提出将INT 制成脂质体后皮肤外用用于治疗皮肤癌,并申请了专利。脂质体包裹的INT 皮肤应用后可以提高INT 在皮肤局部角质层或表皮类脂内的滞留量,延长滞留时间,减少全身吸收,从而增强抗皮肤癌的效果,降低全身性毒性。
脂质体在皮肤局部用药与化妆品领域的应用近30 年来受到了人们的广泛关注,将脂质体分散于水性凝胶基质中制成脂质体凝胶剂是这类制剂的常见应用形式[5-6]。载药脂质体及其凝胶剂的皮肤药物滞留与渗透行为的考察对于阐明制剂的经皮吸收特性是必不可少的,同时有助于探讨脂质体与凝胶的相互作用、制剂皮肤应用后增效减毒的特性以及制剂的作用机制,故本实验研究了INT 脂质体及其凝胶剂的药物离体皮肤滞留与渗透行为。
1 仪器与材料
LC-2130 型高效液相色谱仪、LC-2030 型紫外检测器,上海天美科学仪器有限公司;TT-8D 型药物透皮吸收仪,天津市正通科技有限公司;T-25 型高剪切分散仪,德国易卡公司;Emulsi Flex-C5 型高压均质机,加拿大Avestin 公司;Nicomp 380 CLS型激光散射粒度测定仪,美国Pss 公司;BX51 型光学显微镜,日本奥林巴斯株式会社;透明胶带,北京市鑫田黎明医疗器械有限公司;TDL-80-2S 型低速离心机,上海安亭科学仪器厂;H-600 型透射电子显微镜(TEM),日本HITACHI 株式会社。
INT 原料药,由上海植华生物工程有限公司从峨眉千里光中提取并纯化,质量分数>90%;INT 化学对照品,自制,由INT 原料药经重结晶而得,质量分数>98.0%;Carbopol 934P NF,美国Goodrich公司,批号cc24HBB151;三乙醇胺、尼泊金乙酯、氯仿、氨水、NaCl,分析纯,国药集团化学试剂有限公司;维生素E,1012 IU,西格玛奥德里奇公司;乙腈,HPLC 级,德国Merck 公司;甘油,分析纯,上海麦克林生化科技有限公司;胆固醇,注射级,上海艾韦特医药科技有限公司;大豆磷脂,注射级,磷脂酰胆碱含量大于70%,上海太伟药业有限公司;pH 4.0 柠檬酸缓冲液(CBS 4.0),将0.1 mol/L 柠檬酸溶液与0.2 mol/L 磷酸氢二钠溶液按62∶38 的比例混合配成。HPLC 用水为双蒸水。
新西兰大白兔,雄性,体质量(2.5±0.2)kg,清洁级,由浙江中医药大学动物实验中心提供[合格证号SCXK(沪)2013-0016]。所有动物实验遵循浙江中医药大学动物实验研究中心有关实验动物管理和使用的规定,均符合3R 原则。
2 方法与结果
2.1 样品的制备与表征
进行离体皮肤渗透与滞留考察的样品包括:INT 脂质体、INT 脂质体凝胶剂与INT 普通凝胶剂。
2.1.1 INT 脂质体的制备与表征 按前期研究所得的优化处方与制法[7]制备优化处方脂质体样品。
处方:按制备1000 g 脂质体混悬液成品计,INT 20.5 g、大豆磷脂60.0 g、胆固醇15.0 g、维生素E 3.0 g、CBS 4.0 900 mL、1 mol/L NaOH 水溶液适量。
制法:将INT、大豆磷脂、胆固醇、维生素E溶于1500 mL CHCl3,置于20 L 圆底烧瓶中,50 r/min、60 ℃下旋转蒸发除去CHCl3,在圆底烧瓶瓶壁上形成一层干膜,室温下真空干燥1 d 以除去残余CHCl3,加入处方量CBS 4.0 进行水化,首先振摇使瓶壁上的膜溶散,接着冰箱4 ℃放置8~10 h,最后以高剪切分散仪进行分散(间歇剪切10 次,每次持续时150 s),即得均匀分散的脂质体混悬液(其pH 值在5.29 左右)。
通过调节外水相pH 值,可以利用内、外相pH值梯度将INT 主动导入内水相,即为pH 值梯度载药。在室温、磁力搅拌下,以1 mol/mL NaOH 水溶液适量调节混悬液的pH 值,即得pH 值梯度载药优化脂质体成品,充氮气后4 ℃保存,备用。
为了考察脂质体理化参数对其中包载药物的皮肤渗透与滞留的影响,对优化处方与制法按需要进行适当的调整,制备如下4 份样品:按优化处方制备脂质体混悬液,但未进行pH 值梯度载药,所得样品pH 值较低,包封率较低,且囊泡粒径较大,记为1 号样;按优化处方制备的脂质体混悬液,为了便于比较,与本实验其他考察样品平行制备,所得样品pH 值较高,包封率较高,且囊泡粒径较大,记为2 号样;按优化处方制备脂质体混悬液,但未进行pH 值梯度载药,并进行高压均质(均质次数为8 次,最大压力为14 MPa)使粒径减小,所得样品pH 值较低,包封率较低,且囊泡粒径较小,记为3 号样;对3 号样的制法进行调整,增加pH 值梯度载药步骤(提高2 个单位的pH 值),所得样品pH 值较高,包封率较3 号样高,且囊泡粒径较小,记为4 号样。
采用激光散射粒度测定仪测定上述4 份样品的粒径、ζ 电位,按前期研究中建立的方法进行药物含量与包封率的测定[8],结果见表1。样品1 测得粒径为(349.9±198.0)nm,包封率为12.53%,经过高压均质后的样品3 粒径明显缩小,包封率也相对减小,而采用pH 值梯度载药制备的样品2,粒径为(281.0±49.6)nm,包封率为57.75%,远高于样品1 和样品3。样品4 采用了pH 值梯度载药法并经过高压均质,有部分结晶出现,因此,未进行包封率测定。
表1 4 份不同脂质体样品的理化性质 (±s, n = 3)Table 1 Physicochemical parameters of four different liposome samples (±s, n = 3)

表1 4 份不同脂质体样品的理化性质 (±s, n = 3)Table 1 Physicochemical parameters of four different liposome samples (±s, n = 3)
“-”由于脂质体样品中存在INT 结晶,未进行包封率测定“-” the EE determination wasn’t conducted because of the presence of INT crystal in the liposome sample
样品号 平均粒径/nm ζ 电位/mV 样品形态 INT/(mg·g-1) 包封率/%1 349.9±198.0 1.03 近球形,单、多层共存 19.51 12.53 2 281.0±49.6 1.09 近球形,单、多层共存 18.88 57.75 3 106.1±47.0 0.07 近球形,单层 18.19 7.77 4 118.1±49.3 1.75 近球形,单层,结晶 17.19 -
取少量样品滴于铜网正面,多余样品液用滤纸吸去,滴加少许2%磷钨酸溶液后负染2 min,用滤纸吸去多余染液,自然晾干后,在TEM 下观察并拍摄照片。同时,将少量样品滴于载玻片上,盖上载玻片,用滤纸吸去多余液体,放置在光学显微镜下观察并拍摄照片。TEM 与显微镜下观察脂质体的形态,结果见图1、2。TEM 结果表明,脂质体为类圆形,大小及分布较均匀,颗粒间未有明显的粘连现象。光学显微镜结果可以观察到,脂质体多层的结构。对比3 个样品,可以看出,高压均质后脂质体的粒径明显减小,大粒径脂质体pH 值调节前后的形态无显著变化,小粒径脂质体pH 值调高后有药物结晶析出,在光学显微镜下即可观察到明显结晶,因此,未进行TEM 拍摄。
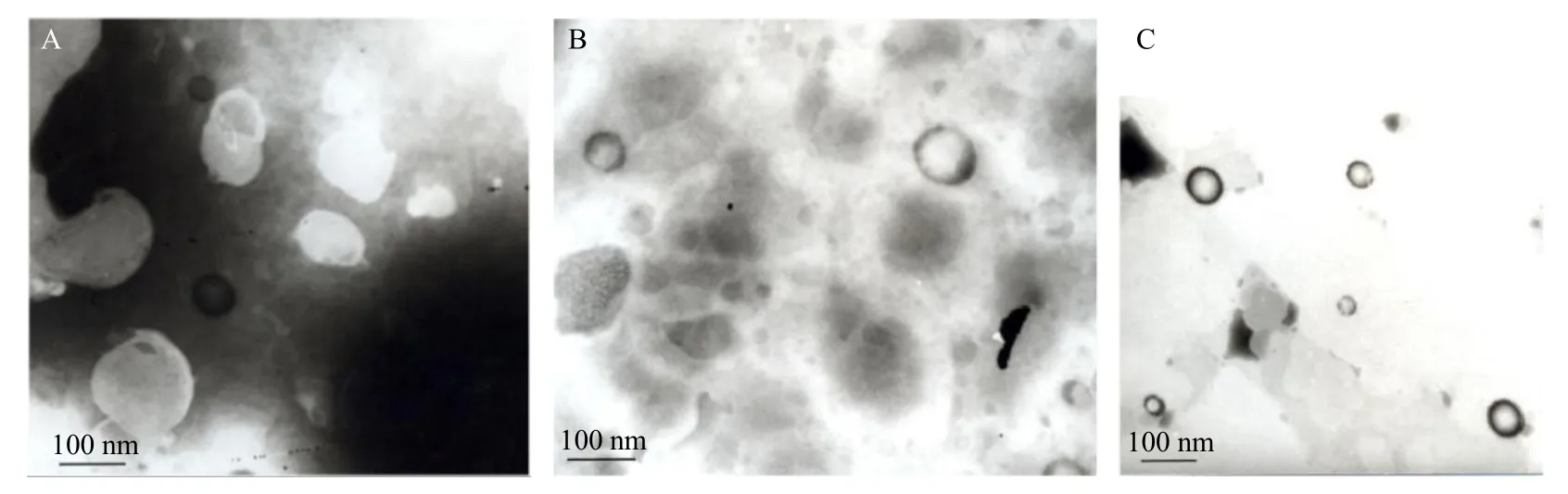
图1 用于离体皮肤渗透试验的脂质体样品1 (A)、2 (B)、3 (C) 的透射电镜图Fig. 1 TEM images of liposome samples 1 (A), 2 (B), 3 (C) for ex vivo skin permeation test
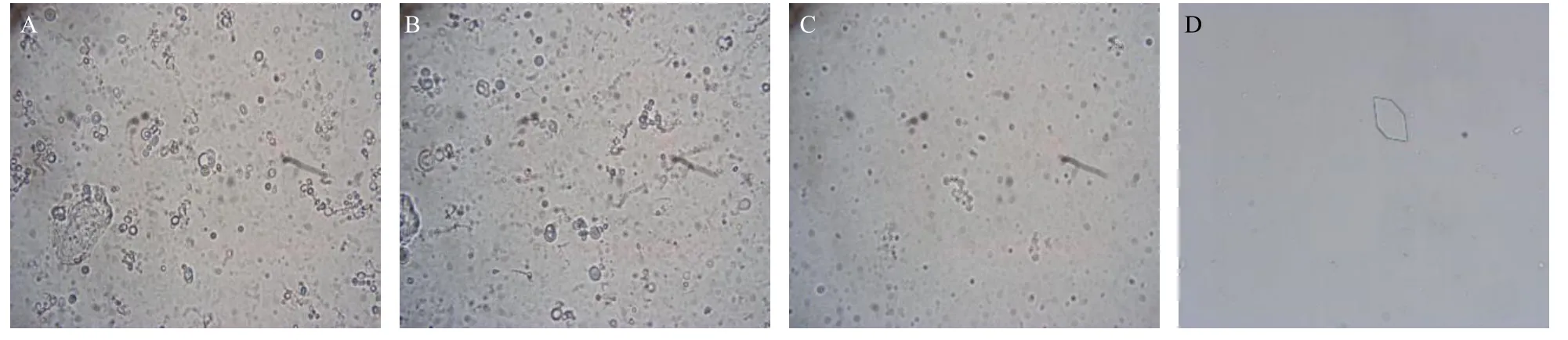
图2 用于离体皮肤渗透试验的脂质体样品1 (A)、2 (B)、3 (C) 与4 (D) 的显微镜照片 (×400)Fig. 2 Microscopic pictures of liposome samples 1 (A), 2 (B), 3 (C) and 4 (D) for ex vivo skin permeation tests (× 400)
2.1.2 INT 脂质体凝胶剂的制备 取优化处方制备的脂质体混悬液,按前期研究所得的处方与制法[7]进行制备。按制备1000 g 脂质体凝胶剂成品计,脂质体混悬液589.3 g、Carbopol 934P 15.0 g、甘油90.0 g、蒸馏水304.5 mL、尼泊金乙酯1.2 g、三乙醇胺适量。取处方量的尼泊金乙酯,加热溶于处方量的蒸馏水中;取处方量的Carbopol 934P,加入甘油润湿,加入溶有尼泊金乙酯的蒸馏水,搅拌均匀,即得INT 脂质体凝胶放置数小时,使Carbopol 934P溶胀呈透明均质状态,慢慢加入脂质体混悬液,边加边搅拌,至均匀,以适量三乙醇胺调节至合适稠度,即得。成品凝胶剂中囊泡分散良好,无聚集;与未加入凝胶的脂质体相比,凝胶中的脂质体圆整度降低、多层脂质体略变少、粒径略减小。测得INT质量分数为11.19 mg/g 的凝胶[7]。
2.1.3 INT 普通凝胶剂的制备及其中INT 含量的测定 按前期研究所得的处方与制法进行制备[9],经含量测定,凝胶剂中INT 的质量分数为11.31 mg/g的凝胶。
2.2 渗透试验接收液中药物INT 的HPLC 测定
2.2.1 色谱条件 参照宋华等[10]建立的色谱条件进行测定。色谱柱为X Terra C18柱(250 mm×4.6 mm,5 μm,Waters);流动相为水-乙腈(70∶30),每1000 毫升流动相中加入冰醋酸1 mL,以氨水将pH 值调至8.6;体积流量0.8 mL/min;检测波长213 nm;柱温为室温。在此条件下,理论塔板数以INT色谱峰计不低于3000。
2.2.2 线性关系考察 以空白渗透试验接收液配制系列质量浓度INT 对照品溶液,按“2.2.1”项下条件进行分析,将INT 的峰面积(A)对质量浓度(C)进行线性回归,回归方程为A=52 771C+3310(r=0.999 9,n=5),可见,INT 在0.499~9.970 μg/mL 内呈良好的线性关系。
2.2.3 精密度试验 取“2.2.2”项下的INT 对照品溶液,进行精密度试验,低、中、高质量浓度的日内精密度分别为1.9%、2.1%、1.5%(n=5),日间精密度分别为2.2%、2.3%、1.7%(n=3),符合要求。
2.2.4 加样回收率试验 以空白渗透试验接收液配制低、中、高质量浓度的INT 对照品溶液,进行回收率试验,结果分别为(101.1±1.3)%、(101.3±1.0)%、(100.6±0.9)%,符合要求。
2.2.5 重复性与稳定性试验 取12 h的离体皮肤渗透试验接收液,在0 时间点按“2.2.1”项下条件进行分析,连续进样5 次,计算RSD,结果为0.8%,说明重复性良好;进而分别在1、2、4、8、12 h 进样分析,计算不同时间点的RSD,结果为1.2%,说明接收液中的INT 在12 h 内稳定。
2.2.6 色谱峰的专属性 对照空白接收液、含药渗透试验接收液与对照品溶液的色谱图表明,空白接收液对测定无干扰,方法专属性符合要求。
2.3 皮肤中滞留药物的HPLC 测定
2.3.1 色谱条件 参照陈思思等[11]建立的色谱条件进行测定。除将流动相改为乙睛-水(23∶77),并用氨水调节其pH 值为7.1 之外,其余同“2.2.1”项下。
2.3.2 皮肤样品的预处理与HPLC 分析 取待测皮肤样品,置于13 mm×65 mm 的有柄研磨器中,加入2 mL 氨水作为研磨介质进行研磨,直至将皮肤磨成均匀的浆状,再加入1 mL 氨水,涡旋混合15 s,用醋酸乙酯提取3 次,各次醋酸乙酯的用量分别为3、2、2 mL,每次提取涡旋30 s、2500 r/min 离心5 min,合并各次醋酸乙酯提取液,氮气吹干,以乙腈-水(1∶1)混合液200 μL 溶解残渣后,吸取20 μL 进样进行HPLC 分析。
2.3.3 色谱峰的专属性 取离体皮肤渗透实验中药物应用720 min 后的皮肤半块、等面积空白皮肤、等面积空白皮肤加一定量的INT 对照品溶液,分别按上述方法进行样品预处理与HPLC 分析,色谱图见图3-A~C,由图可见,INT 的出峰时间在10.7 min,皮肤中的内源性成分对INT 的含量测定无干扰,方法专属性符合要求。
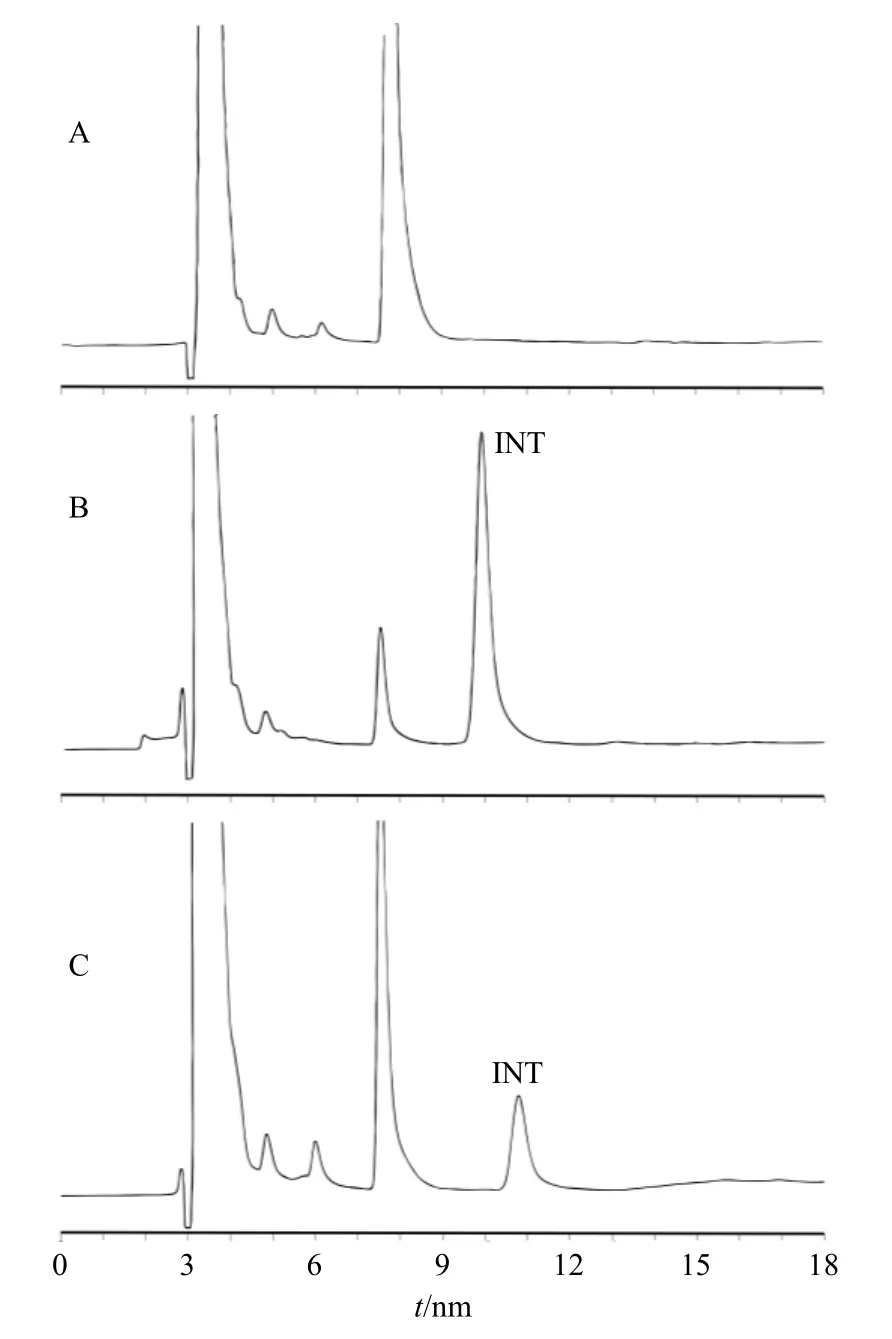
图3 HPLC 测定皮肤中INT 的色谱峰专属性试验色谱图Fig. 3 Chromatographic peak specificity test chromatograms for determination of INT in skin by HPLC
2.3.4 线性关系考察 取空白皮肤1 cm2,加入不同体积的一定质量浓度INT 对照品水溶液(其中INT 总药量依次为0.525、2.100、4.200、10.500、15.750、21.000、31.500 μg)。按上述方法对样品进行预处理与HPLC 分析,进样后记录INT 对应的峰面积(A),将峰面积对INT 总量(m)进行线性回归,回归方程为A=203 992m-24 120(r=0.999 8,n=7),可见,INT 在0.525~31.500 μg 内呈良好的线性关系。另外,经测定本法的最低检测限为0.05 μg(S/N≥3)。
2.3.5 回收率、精密度与提取回收率试验 取空白皮肤1 cm2,加入不同体积的一定质量浓度INT 对照品水溶液(其中INT 总药量依次为0.525、4.200、31.500 μg)。按“2.3.2”项下方法对样品进行预处理,并按“2.3.1”项下方法进行HPLC 分析,记录INT对应的峰面积,各个加药量的空白皮肤样品分别在1 d 内连续测定5 份,求得日内精密度,并由标准曲线计算相对回收率,各个加药量的空白皮肤样品分别在5 d 各测1 份,求得日间精密度,结果见表2。可见,方法精密度与准确性良好。

表2 日内、日间精密度与相对回收率试验结果 (n = 5)Table 2 Within-day and inter-day precision and relative recovery (n = 5)
另将INT 对照品水溶液直接进样,测得相应峰面积,以此为参照,计算空白皮肤中3 种不同加药量(分别为0.525、4.200、31.500 μg)样品的提取回收率分别为79.97%、82.09%、78.48%,平均值为80.18%,符合生物样本含量测定中对提取回收率的要求。
2.4 离体皮肤的制备
取健康新西兰大白兔(体质量2.5~3.0 kg),剪去耳背部的毛。以过量乙醚麻醉致死后,取下耳背部的皮肤,除去皮下组织,洗净,在生理盐水中浸泡后展平,夹在两玻璃板间,塑料袋密封包装后置于冰箱低温保存,备用。
2.5 离体皮肤渗透
2.5.1 实验过程 采用Franz 扩散池进行实验[12]。取冰冻兔耳耳背皮肤,室温放置解冻,剪取合适大小(约1.4 cm×1.4 cm)的正方形皮肤样品,适当拉力下展平,固定于供给池与接收池的磨口对接处,角质层面向供给池,有效渗透面积为0.785 cm2(按扩散池内口直径1.00 cm 计算得到)。
分别将成品INT 脂质体凝胶剂0.25 g、优化脂质体混悬液0.15 g 与INT 普通凝胶剂0.25 g 均匀地涂布于皮肤角质层上,实验中供给池保持非封闭状态。接收池中加入6.0 mL 生理盐水(用煮沸适当时间放冷后的蒸馏水配制)作为接收液,37 ℃恒温水浴浸没接收池,400 r/min 定速搅拌接收液,于15、30、60、120、180、240、360、480、600、720 min从接收液中取样0.25 mL,同时向接收池中补加等量同温的生理盐水。
从所取样品液中吸取20 μL 进样,HPLC 测定其中药物浓度。考虑取样药物损失,计算各时间点的累积药物透过量,将透过量对时间作图,由透过量曲线后段的直线部分求算药物稳态渗透速率。每个样品平行测定4 份。不同理化参数的4 份脂质体样品同上法测定,投药量均为0.15 g。
2.5.2 实验结果 不同样品中INT 透过皮肤的稳态渗透速率见表3,由于所有样品中药物渗透速率均较小,因此测量值的相对误差均较大,再加上皮肤本身的差异,所以表中的数据波动较大。从表中可以看出,这些样品总的特征是透过量小,其中脂质体凝胶剂的稳态渗透速率为(1.863±1.668)μg/(h·cm2),证明它具有减毒的特性。
表3 不同样品中INT 的稳态渗透速率 (±s, n = 4)Table 3 Steady state permeation rate of INT in different samples (±s, n = 4)
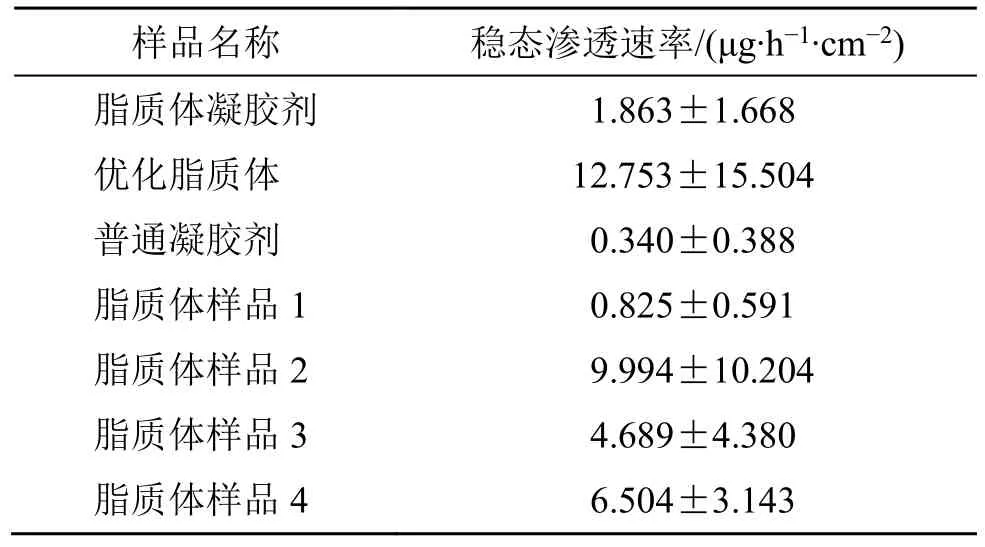
表3 不同样品中INT 的稳态渗透速率 (±s, n = 4)Table 3 Steady state permeation rate of INT in different samples (±s, n = 4)
样品名称 稳态渗透速率/(μg·h-1·cm-2)脂质体凝胶剂 1.863±1.668优化脂质体 12.753±15.504普通凝胶剂 0.340±0.388脂质体样品1 0.825±0.591脂质体样品2 9.994±10.204脂质体样品3 4.689±4.380脂质体样品4 6.504±3.143
比较表3 中的数据可以发现,稳态渗透速率上存在大小关系如下:优化脂质体>脂质体凝胶剂>普通凝胶剂[经方差分析,F=2.26<F1-0.05(2,9)=4.26,无显著性差异];不同理化参数的脂质体的透过速率之间的大小关系如下:样品2>样品4>样品3>样品1[经方差分析,F=1.75<F1-0.05(3,12)=3.49,无显著性差异]。
2.6 离体皮肤滞留
2.6.1 实验过程 同“2.5.1”项下操作,720 min 后从扩散池之间取下皮肤,将皮肤表面残存的药物用水清洗掉,用吸水纸吸干皮肤表面。将皮肤展平于玻璃板上,将有效扩散皮肤部分切成同样大小的两半,其中一半用透明胶带在角质层侧粘贴并剥离30次以去除皮肤的角质层[15],将两半皮肤分别按“2.3”项下方法测定INT 含量。每个样品平行测定4 份。
2.6.2 不同样品中INT 的皮肤滞留 不同样品应用后皮肤中INT 的滞留量测定结果见表4,去角质层的皮肤中的滞留量测定结果见表5,根据以上数据,进一步求得角质层药物滞留量占全皮中药物滞留量的百分比,结果见表6。
表4 不同样品应用12 h 后全皮肤中INT 的滞留量 (±s,n = 4)Table 4 Deposited amount of INT in whole skin 12 h after application of different samples (±s, n = 4)
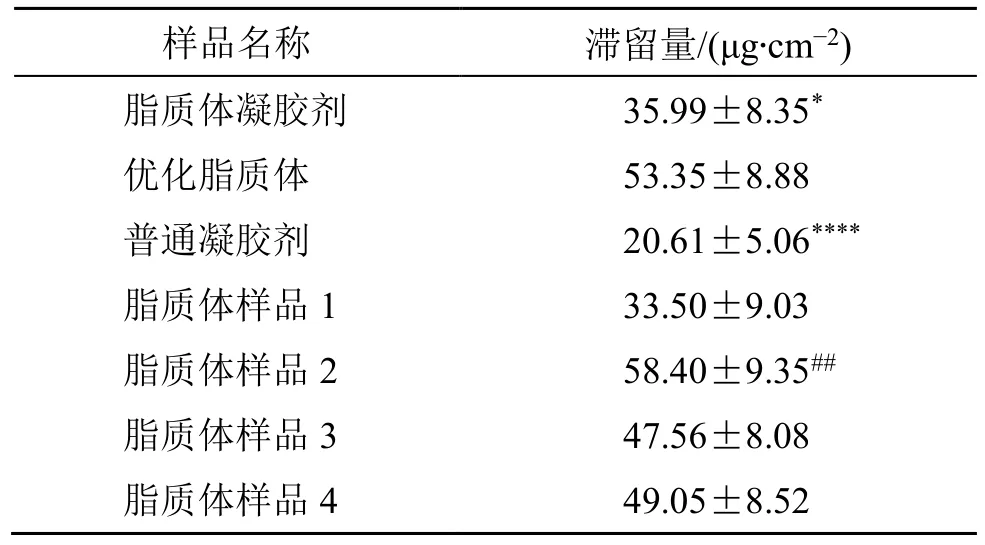
表4 不同样品应用12 h 后全皮肤中INT 的滞留量 (±s,n = 4)Table 4 Deposited amount of INT in whole skin 12 h after application of different samples (±s, n = 4)
与优化脂质体比较:*P<0.05 ****P<0.000 1;与脂质体样品1 比较:##P<0.01*P < 0.05 ****P < 0.000 1 vs optimized liposome; ##P < 0.01 vs liposome sample 1
样品名称 滞留量/(μg·cm-2)脂质体凝胶剂 35.99±8.35*优化脂质体 53.35±8.88普通凝胶剂 20.61±5.06****脂质体样品1 33.50±9.03脂质体样品2 58.40±9.35##脂质体样品3 47.56±8.08脂质体样品4 49.05±8.52
表5 不同样品应用12 h 后角质层下皮肤中INT 的滞留量(±s, n = 4)Table 5 Deposited amount of INT in skin beneath stratum corneum 12 h after application of different samples (±s,n = 4)
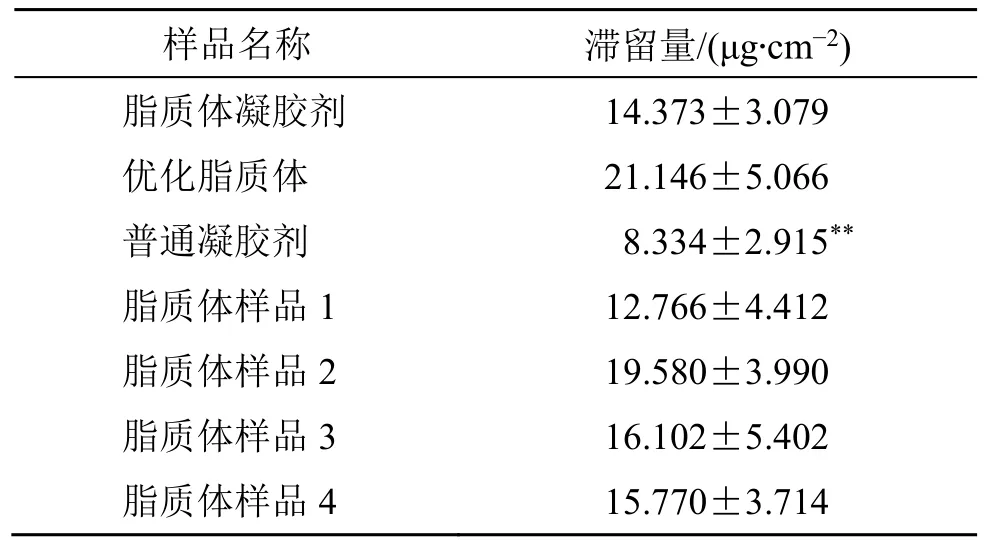
表5 不同样品应用12 h 后角质层下皮肤中INT 的滞留量(±s, n = 4)Table 5 Deposited amount of INT in skin beneath stratum corneum 12 h after application of different samples (±s,n = 4)
与优化脂质体比较:**P<0.01**P < 0.01 vs optimized liposomes
样品名称 滞留量/(μg·cm-2)脂质体凝胶剂 14.373±3.079优化脂质体 21.146±5.066普通凝胶剂 8.334±2.915**脂质体样品1 12.766±4.412脂质体样品2 19.580±3.990脂质体样品3 16.102±5.402脂质体样品4 15.770±3.714
表6 不同样品应用12 h 后在角质层中INT 的滞留量占全皮的百分比 (±s, n = 4)Table 6 Percentage of deposited amount of INT in stratum corneum to that in whole skin 12 h after application of different samples (±s, n = 4)
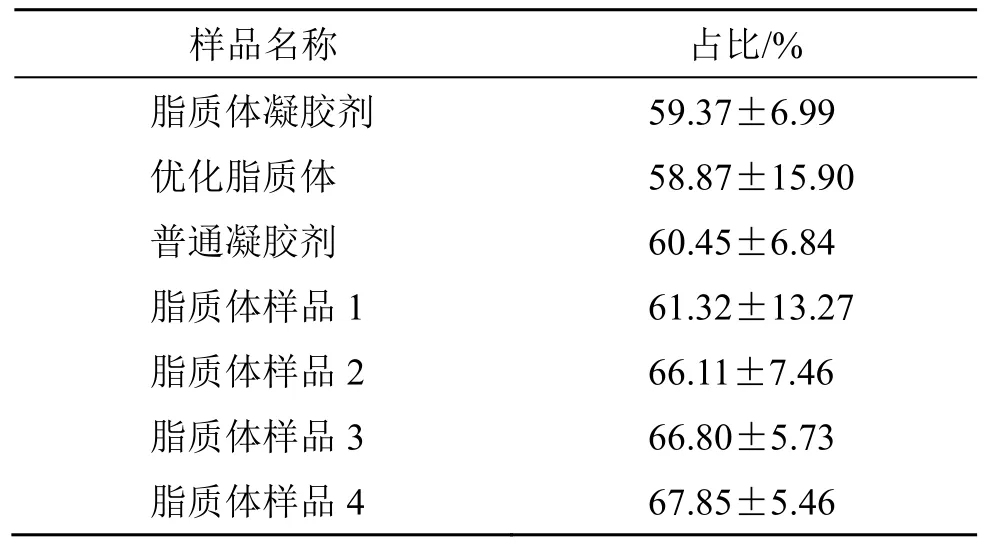
表6 不同样品应用12 h 后在角质层中INT 的滞留量占全皮的百分比 (±s, n = 4)Table 6 Percentage of deposited amount of INT in stratum corneum to that in whole skin 12 h after application of different samples (±s, n = 4)
样品名称 占比/%脂质体凝胶剂 59.37±6.99优化脂质体 58.87±15.90普通凝胶剂 60.45±6.84脂质体样品1 61.32±13.27脂质体样品2 66.11±7.46脂质体样品3 66.80±5.73脂质体样品4 67.85±5.46
由表4 可以看出,不同制剂应用12 h 后皮肤中滞留量的大小关系如下:优化脂质体混悬液>脂质体凝胶剂>普通凝胶剂(2.59∶1.75∶1)。对表4 中制剂①脂质体凝胶剂、制剂②优化脂质体混悬液与制剂③普通凝胶剂应用后皮肤中药物滞留量进行方差分析,结果表明,不同制剂对药物皮肤滞留量有极显著的影响[F=18.48>F1-0.01(2,9)=8.02]。两两间多重比较的结果表明,制剂②与③之间有极显著性差异(P<0.000 1,q1-0.01=5.43),①与②之间有显著性差异(P<0.05,q1-0.05=3.95),①与③之间无显著性差异。另外,对表4 中脂质体样品1~4 应用12 h 后皮肤中的药物滞留量进行方差分析,结果表明,脂质体理化参数不同对药物皮肤滞留量有显著的影响[F=5.51>F1-0.05(3,12)=3.49]。两两间多重比较的结果表明,样品1 与2 之间有极显著性差异(P<0.01,q1-0.01=5.50),其余两两之间均无显著性差异(P>0.05,q1-0.05=4.20)。样品2 为经过pH 值主动载药的大粒径脂质体,包封率最高,上述实验结果证明高包封率脂质体有利于INT 的皮肤转运。
从表5 可以看出,不同制剂皮肤应用12 h 后,在角质层下皮肤中的滞留量的大小关系如下:优化脂质体混悬液>脂质体凝胶剂>普通凝胶剂(2.54∶1.72∶1)。对表5 中制剂①脂质体凝胶剂、制剂②优化脂质体混悬液与制剂③普通凝胶剂应用12 h 后角质层下皮肤中的药物滞留量进行方差分析,结果表明,不同制剂对角质层以下皮肤中药物滞留量有极显著的影响[F=11.30>F1-0.01(2,9)=8.02]。两两间多重比较的结果表明,制剂①与②,①与③之间均无显著性差异(P>0.05,q1-0.05=3.95);②与③之间有极显著性差异(P<0.01,q1-0.01=5.43)。对比表4 与表5 中还可以看出,3 种制剂应用后在角质层下皮肤中的滞留量的大小比例关系与在全皮中滞留量的大小比例关系基本一致。
对表6 中制剂①脂质体凝胶剂、制剂②优化脂质体混悬液与制剂③普通凝胶剂应用12 h后角质层药物滞留量占全皮中药物滞留量的百分数进行方差分析,结果表明,不同制剂对角质层药物滞留量占全皮中的百分比均无显著性影响[F=0.024<<F1-0.05(2,9)=4.26]。占比均在60%左右,表明进入皮肤的药物在角质层中的分布较下层皮肤多一些。
3 讨论
本实验建立了一种皮肤中INT 含量测定的HPLC 方法,该法准确,精密。测定中皮肤前处理采用物理研磨法进行皮肤匀浆,而未使用强烈腐蚀性的化学物质(如高氯酸[13]等),有利于待测成分的稳定。多数报道表明,实验动物中家兔皮肤的渗透性最大,故很难作为药物人体透皮吸收预测的模型,而正是由于吸收快的性质,家兔可以用来指示药物的皮肤毒性[14]。故本实验采用家兔皮肤作为渗透试验皮肤。此外,有文献报道[15]用硫化钠溶液处理皮肤以脱去皮肤上的毛,但硫化钠可能对皮肤结构产生影响,故本实验未采用。
离体皮肤渗透实验的结果表明,普通凝胶剂、脂质体与脂质体凝胶剂的皮肤透过量均较小,透过速率上大小关系为优化脂质体>脂质体凝胶剂>普通凝胶剂;不同理化参数的脂质体的透过速率之间的大小关系为pH 值梯度载药的大粒径脂质体>pH值梯度载药的小粒径脂质体>未经pH 值梯度载药的小粒径脂质体>未经pH 值梯度载药的大粒径脂质体。
由此可以推测,脂质体对药物透过皮肤的转运有一定促进作用,而这种作用的前提是脂质体与皮肤直接接触,故脂质体混悬液的药物渗透速率最大,脂质体凝胶剂中由于凝胶限制了脂质体与皮肤的相互作用,所以药物转运速率明显下降,而普通凝胶剂缺乏这种作用,故药物渗透速率最小。按照透析袋中释放实验的结果,凝胶剂与脂质体凝胶剂药物释放较快,可以推测,两者应用于皮肤后可以在皮肤表面产生较高的游离药物浓度,而脂质体混悬液在皮肤表面产生的游离药物浓度较低[9],然而,离体皮肤渗透的结果却表明前两者药物皮肤渗透速率较脂质体混悬液明显低,由此进一步证明了脂质体对药物转运的促进作用。
关于脂质体促进药物皮肤转运有如下4 种可能的机制[16-27]。第一,通过脂质体的脂质双分子层与皮肤脂质融合直接将脂质双分子层中的药物转运到皮肤,第二,药物由脂质体的脂质双分子层分配到皮肤角质层脂质,第三,脂质体扰乱皮肤角质层中的脂质的有序排列使游离药物的皮肤渗透增加,第四,脂质体以整体的形式进入皮肤来实现药物的皮肤转运。大量的研究[17-18,20,25-26]已将第4 种机制否定,即,胶态脂质体不能进入完整的皮肤。对照优化脂质体、脂质体凝胶剂、普通凝胶剂的皮肤渗透速率,不能对INT 脂质体促进药物皮肤转运的机制作出明确的推测,可能是前3 种机制兼有或其中某1 种或2 种。对比不同理化参数的脂质体的透过速率可以看出,进行pH 值梯度载药的脂质体的透过速率较大,其中大粒径的脂质体较小粒径的脂质体透过速率大。因为提高外水相pH 值之后,药物包封率提高;大粒径脂质体因为包封体积大,所以包封率高,这2 点都有利于增加通过脂质体与皮肤相互作用提高的药物进入皮肤的量,这一结果进一步支持了脂质体促进药物皮肤转运的结论,同时可以探明其机制主要为前2 种,因为如果是第3 种机制,药物皮肤转运量与脂质体的药物包封率不会有很大关系。
由表4 可知,不同制剂应用12 h 以后皮肤药物滞留量的大小关系如下:优化脂质体>脂质体凝胶剂>普通凝胶剂(2.59∶1.75∶1)。由此推断,通过脂质体与皮肤的相互作用可以促进药物进入皮肤,然而,凝胶在一定程度上阻碍脂质体与皮肤的相互作用的发生,从而减小了脂质体促进药物进入皮肤的作用。但是,从实际制剂应用的方便和制剂的稳定性方面考虑,脂质体凝胶剂是有利的。使用体温溶胶化的凝胶或者触变胶有望将单纯脂质体在皮肤滞留方面的优势与脂质体凝胶剂在应用方便、提高脂质体稳定性等方面的优势集中在一起。
由表5 可知,不同制剂皮肤应用12 h 后,在角质层以下皮肤中的滞留量的大小关系如下:优化脂质体>脂质体凝胶剂>普通凝胶剂(2.54∶1.72∶1)。3 种制剂应用后在角质层以下皮肤中的滞留量的大小比例关系与在全皮中滞留量的大小比例关系基本一致,并且,角质层药物滞留量占全皮中药物滞留量的百分数无显著差异,均在60%左右,由此推断,脂质体的皮肤药物转运机制是角质层与脂质体的脂质之间直接分配,而不是脂质融合,因为如果有融合机制存在,脂质体应用后的皮肤角质层的药物浓度会显著增大,上述比例关系的一致性就不大可能了。因此,脂质体与皮肤的相互作用仅局限于皮肤的角质层的表面,进入角质层的药物向皮肤更深层次的转运完全与药物自身的性质有关,而与脂质体无关。此外,对表5 中脂质体样品1~4 应用12 h 以后角质层以下皮肤中的药物滞留量进行方差分析,结果表明,脂质体理化参数不同对角质层以下皮肤药物滞留量无显著性的影响[F=1.59<<F1-0.05(3,12)=3.49]。按上述进入角质层的药物向皮肤更深层转运为自主过程,仅与药物本身性质有关,所以根据不同理化性质的脂质体应用后在全皮中的滞留量不同,可以推断在角质层下皮肤中的滞留量也是不同,但是由于这一层中药物滞留量较小,所以统计结果未显现显著性差异。
样品2 为经过pH 值主动载药的大粒径脂质体,包封率最高,上述实验结果证明这种脂质体最有利于INT 的皮肤转运。前面已有推断,脂质体与皮肤之间的直接药物分配是INT 脂质体促进药物进入皮肤的机制,包封率高的脂质体可以将更多的药物运送到角质层也支持了这一推断。其他,1 号样为未进行pH 值梯度载药的大粒径脂质体,由于包封率低,皮肤药物滞留量明显较少;3 号样为未进行pH值梯度载药的小粒径脂质体,包封率也较低,但其皮肤滞留量较1 号样有所增加,这可能是因为小粒径脂质体在促进药物进入皮肤方面存在着其他的机制(如通过毛囊转运);4 号样为经过pH 值梯度载药的小粒径脂质体,由于包封容积小,有药物结晶存在,该样品产生相对较高的皮肤滞留量一方面可能是因为单独脂质体上载药量较高(脂质体内、外水相药物浓度均已达到饱和),另一方面可能也是由于其他机制的存在。
利益冲突所有作者均声明不存在利益冲突
