线粒体功能障碍与阿尔茨海默病的相关性研究
2023-05-31叶群英综述王艳云罗红波审校
叶群英 综述 王艳云,罗红波 审校
遵义医科大学第五附属(珠海)医院神经内科,广东 珠海 519100
阿尔茨海默病(Alzheimer's disease,AD)是一种以进行性认知功能障碍和行为学损害为主要临床表现的神经变性疾病。该病占痴呆症患者总数的60%~80%[1-2]。据统计,截止目前包括AD 在内的全球痴呆患者约5 000 万,估计到2050年将增长2 倍,并且基于AD生物学特性,甚至可能增加3倍[3]。但AD的病因是复杂、多元的,关于AD的具体发病机制尚不清楚。目前较为公认的机制假说包括:淀粉样蛋白假说、Tau蛋白假说、胆碱能假说、APOE4 基因学说及氧化应激假说、线粒体功能障碍假说等[4]。目前,AD治疗药物的研发主要围绕着“淀粉样蛋白假说、Tau蛋白假说”,但仍不能有效延缓或者遏制AD的疾病进程,因此充分研究AD的发病机制,找到合理的治疗靶点,寻求有效的药物,仍是当前亟待解决的问题。近年来越来越多研究表明,线粒体功能障碍在AD发病机制中发挥的重要作用,对线粒体方面相关机制的研究已成为研发AD靶向治疗药物的热点[5-7]。本文就线粒体能量代谢、Ca2+稳态、氧化应激、线粒体动力学、线粒体DNA、线粒体自噬等方面,对线粒体功能障碍与AD的相关性研究进行探讨,以期为AD的临床研究与治疗方向提供科学依据。
1 线粒体结构及功能
线粒体是一种真核生物的细胞器,为双层单位膜构成的封闭囊状结构,分为基质、内膜、膜间隙、外膜。内膜上有氧化磷酸化相关的蛋白质,包括呼吸链(复合物Ⅰ、Ⅱ、Ⅲ、Ⅳ)、ATP 合成酶及腺苷转运体等[8]。内源性自由基主要由电子传递链产生,其可损伤蛋白质、DNA和脂质。此外,线粒体基质有着完整的转录与翻译体系,其包括线粒体DNA、tRNA、tRNA、70S型核糖体、DNA 聚合酶、氨基酸活化酶等。线粒体是机体的“能量工厂”,其通过合成ATP,调控Ca2+通道,调节氧化应激(oxidative stress,OS)及细胞凋亡,维持着机体能量代谢[9]。同时,线粒体又可通过分裂、融合、转运与自噬维持自身的动态平衡[10]。
2 线粒体功能障碍与AD的相关性
2.1 线粒体能量代谢障碍与AD的相关性 人脑占机体体质量的2%~3%,在耗氧量方面,约占机体的20%[11],且ATP 是大脑唯一的能量使用形式。绝大部分ATP是依赖于线粒体内膜上的氧化磷酸化的生成,氧化磷酸化需要电子传递链和ATP 合成酶的参与[12]。因此,复合体Ⅰ~Ⅳ和ATP 合成酶的活性改变或基因表达下降将直接影响着线粒体能量代谢。研究发现线粒体功能障碍导致的神经元“饥饿”与进行性认知和运动退化有着密切的关系[13]。Terni等[14]发现,与正常人相比,在AD 患者的早期(Braak Ⅰ/Ⅱ期,MCI 发作前),线粒体ATP 合成酶的α亚单位在内嗅皮质中被HNE修饰,可使ATP合成酶的活性下降约30%。Chou等[2]发现,在AD 小鼠(3xTg-AD)皮质中进行的蛋白质组学测定显示,与对照组相比,AD小鼠中ATP合成酶的β亚单位的表达是增加。根据上述可以推测,通过改变ATP 合成酶的α亚单位或者β亚单位表达可以影响ATP 酶的活性,从而来影响线粒体的能量代谢,但其具体的机制尚未阐明。与对照组比较,AD 晚期患者编码线粒体电子传递链亚基的核基因在后扣带皮层、海马CA1 区、颞中回和内嗅皮层中的表达是减少的[15]。在AD 中,线粒体功能障碍也可通过抑制线粒体呼吸链复合物Ⅰ、Ⅲ和Ⅳ的表达,从而影响其能量代谢[16]。因此,可以进一步推测通过靶向性干预ATP合成酶或者其他复合体来提高ATP的产生,达到恢复其正常能量代谢的目的。
2.2 钙离子稳态失衡与AD的相关性 钙离子稳态(calcium homeostasis)是细胞中大部分生化反应的基础,钙离子主要在内质网储存,在线粒体中发挥作用,调节细胞基本功能[17]。一方面,线粒体Ca2+摄取主要是受线粒体外膜上的电压依赖性阴离子通道(VDAC)和线粒体内膜上的线粒体Ca2+单转运蛋白复合物(MCU)调节;另一方面,线粒体Ca2+排出主要通过线粒体内膜的Na+/Ca2+/Li交换器(NCLX)和Ca2+/H+交换器(CHE/Letm1)调节[18-19](图1)。研究发现,β淀粉样蛋白(Aβ)聚集体通过调节MCU提高线粒体的Ca2+摄取,致使线粒体Ca2+超载;同时用特异性抑制剂Ru360 阻断MCU 可以避免Ca2+超载[20]。研究发现,在AD 患者及3×Tg AD 小鼠中的NCLX 表达下降,造成线粒体Ca2+排出受阻,使得线粒体Ca2+超负荷,迫使开放线粒体通透性转变孔(MPTP),致使神经元凋亡,促进AD 的发展;同时恢复或者提高NCLX的表达能够促使线粒体Ca2+正常排出,避免了线粒体Ca2+超负荷运转、细胞凋亡,进而改善AD 病理和认知障碍[21]。上述证据表明在AD早期可通过靶向性调节线粒体Ca2+的摄入或排出,可避免线粒体Ca2+超载,恢复线粒体Ca2+稳态,从而避免神经元凋亡,达到预防治疗AD的目的。
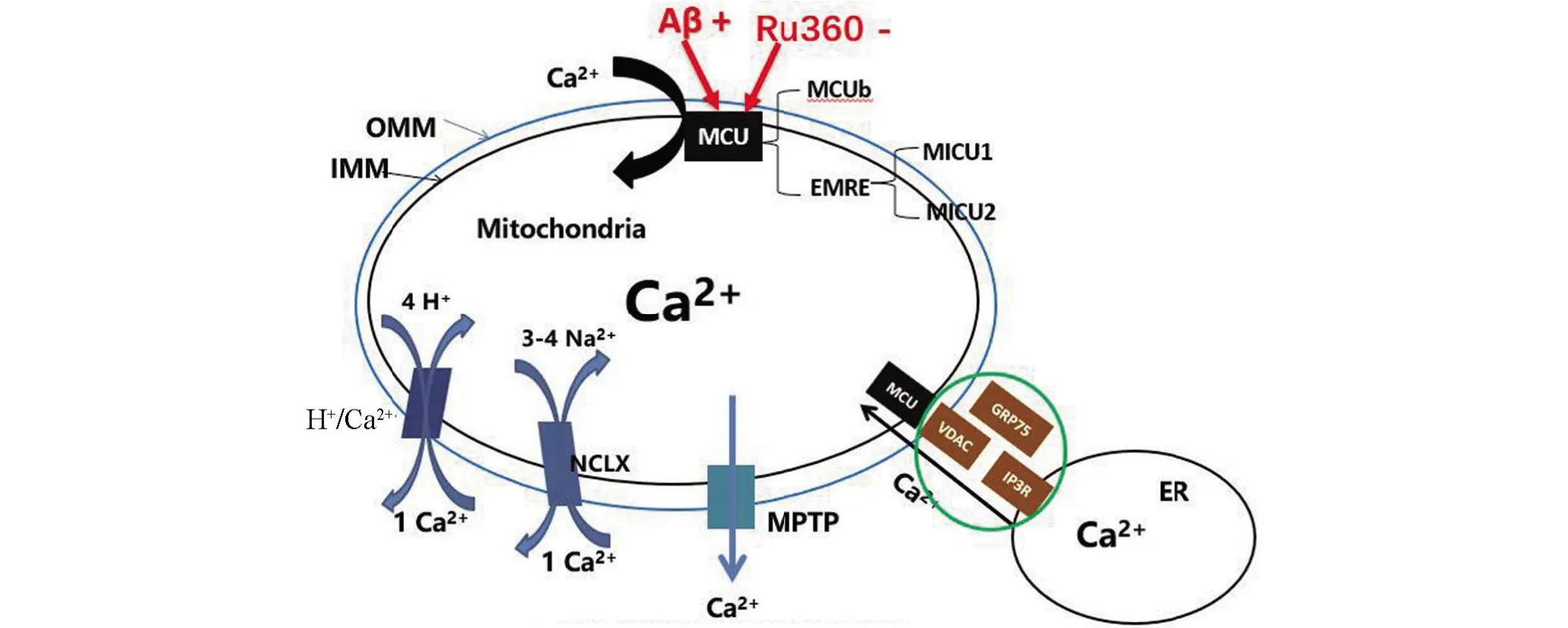
图1 钙离子通道稳态及钙超载示意图Figure 1 Schematic diagram of calcium homeostasis and calcium overload
2.3 氧化应激与AD的相关性 氧化应激(oxidative stress,OS)是指体内活性氧(reactive oxygen species,ROS)或活性氮(reactive nitrogen species,RNS)的产生和蓄积与机体对其清除能力的不对等所引起的现象[22]。线粒体是内源性ROS的主要产生场所[23]。在线粒体正向电子传递中,呼吸链上的复合物Ⅰ、Ⅱ和Ⅲ产生不可避免的电子泄漏,泄漏的电子与氧气相互作用形成超氧阴离子(superoxide anion radical,O2-或过氧化氢(H2O2)[24];在线粒体反向电子传递中,随着质子动力上升,可驱动电子通过辅酶Q从复合物Ⅱ转移到复合物Ⅰ,将NAD+还原成NADH,导致NADH/NAD+比值增大,使线粒体处于过氧化状态,从而极大提高O2-含量,升高了线粒体ROS水平[25]。Mclellan 等[26]采用多光子成像技术对AD动物脑内Aβ沉积物进行成像,发现自由基产生的荧光分布在淀粉斑周围。Lovell 等[27]利用Cu2+和Fe2+发生经典的芬顿反应造成原代大鼠皮层神经元的氧化应激,使得Tau蛋白的构象发生改变,并使蛋白激酶C 的α、β异构体失活,而蛋白激酶C 调节GSK-3β,诱导Tau 蛋白磷酸化,促使神经元凋亡。由于线粒体DNA离线粒体内膜更近,故更易遭到自由基的破坏,同时缺乏了组蛋白保护与损伤修复系统,故比nDNA更易发生突变[28]。上述证据表明线粒体氧化应激可通过上调Aβ表达、上调P-Tau 蛋白表达及损伤线粒体DNA等生物分子,进一步促进了AD的发生发展。由此,可以认为通过干预线粒体氧化应激靶向性治疗AD具有一定的可行性。
2.4 线粒体动力学失衡与AD的相关性 线粒体动力学是指线粒体通过分裂和融合的动态转化,来维持自身稳态的过程,其受多种相关蛋白的调控。包括线粒体有丝分裂蛋白1(动力蛋白相关蛋白1)、视神经萎缩相关蛋白1 (OPA1)和线粒体融合蛋白1 和2(mitofusion1/2)。线粒体融合主要由Opa1、Mfn1 和Mfn2共同调控,Opa1位于线粒体内膜上,推进线粒体内膜融合;Mfn1 位于线粒体外膜,促进线粒体外膜融合;Mfn2在线粒体外膜和内质网上促进线粒体外膜融合及协调线粒体-内质网交互作用[29-30]。线粒体转运系统是由细胞骨架(微丝、微管)、马达蛋白(驱动蛋白、动力蛋白、肌球蛋白)及衔接蛋白(Miro/Milton 等)组成[31-33]。研究发现,在哺乳动物中,Drap1、MFF、MiD49和MiD51共同调控线粒体分裂,Drp1在某些特定细胞信号传导下由细胞质招募到线粒体外膜,同时与线粒体外膜上的线粒体分裂因子(MFF)、线粒体动力蛋白(MiD49h、MiD51)结合。Drp1 通过环状结构寡聚水解GTP提供能量使该环状结构收缩,离断双层膜结构,以致线粒体分裂[34-35]。有研究发现,将3 个月、6个月、9 个月、12 个月的APPsw/PS1dE9 基因鼠与同月龄的C57BL/6 小鼠相比,从第3 个月开始基因鼠海马组织中的线粒体形态出现改变,并伴有Opa1 和Mfn2及Drap1 的改变[36]。同时有研究发现,在Aβ及Tau 蛋白出现之前可观察到轴突肿胀及线粒体轴突运输减少,这揭示线粒体运输功能障碍可能也是AD 的早期生物学标记物[37]。根据上述研究,可以推测线粒体分裂、融合及运输的改变可能是早期AD发展的标志,其通过靶向性调控Opa1、Mfn2、Drap1等蛋白的表达,可阻止AD的进一步发展,达到逆转或延缓AD的可能。
2.5 线粒体DNA异常与AD的相关性 人类的线粒体DNA 共有37 个基因,分别编码22 个tRNA、2 个rRNA 及13 种氧化磷酸化酶复合体亚基(NADH-CoQ还原酶的7个亚基、泛醌-细胞色素C还原酶的1 个亚基、细胞色素C氧化酶的3个亚基及ATP合成酶的2个亚基)[38]。由于线粒体DNA离线粒体内膜更近,更易受到自由基的破坏,同时其缺乏组蛋白保护和损伤修复机制,故比nDNA更易发生变异[39]。线粒体DNA中唯一的非编码区是D环区,负责线粒体DNA复制与转录,是最易受氧化损伤而发生变异的区域[40]。在AD等神经变性疾病中,线粒体DNA拷贝数水平变化与线粒体DNA甲基化水平的动态调控和线粒体DNA复制转录因子的表达下降有关[41]。研究发现,在AD 患者的额叶皮层中,D 环区突变显著增加了63%,线粒体DNA 拷贝数同时下降了50%[42]。研究显示,早期AD患者大脑内嗅皮层中,D环区甲基化水平明显增加,且早期甲基化水平高于晚期[43]。Sheng 等[44]研究发现,AD 患者海马组织中线粒体转录因子A 水平下降43%,并引起mt DNA 拷贝数的减少。根据以上证据可以认为,D环区突变、甲基化及线粒体DNA复制转录因子下降均可导致线粒体DNA异常,致使线粒体DNA拷贝数减少,从而导致线粒体功能障碍。因此,可以以挽救线粒体DNA 异常、恢复线粒体DNA 拷贝数水平、避免线粒体功能障碍来阻止AD的发生发展。
2.6 线粒体自噬异常与AD的相关性 线粒体自噬是一种控制线粒体质量的选择性自噬途径,其通过双膜自噬体结构吞噬受损或去极化的线粒体,与溶酶体融合,完成受损或去极化线粒体的降解[45]。有研究发现,线粒体自噬主要受同源磷酸酶张力蛋白诱导的蛋白激酶1(PINK1)/Parkin信号通路的调节[46]。PINK1是位于线粒体外膜的丝氨酸/苏氨酸激酶;Parkin是位于胞质内的E3泛素连接酶。其大致过程为:在健康机体中,某一因素致使线粒体膜电位下降时,PINK1可迅速聚集并招募Parkin 至线粒体外膜上进行激活,活化的Parkin 可介导受损线粒体外膜蛋白泛素化,促进自噬小体初始化形成,并诱导微管相关蛋白1 轻链3(LC3)至LC3 相互作用区(LC3-interation region,LIR),促使受损的线粒体形成线粒体自噬体,再与溶酶体融合后加速受损线粒体的降解及清除[47-48]。Ye 等[13]发现,在AD患者大脑中Parkin水平会随着Braak分期的增加而逐渐下降。还有研究发现,与健康对照组相比,AD 患者脑组织中线粒体自噬的基础水平低于50%[49]。新的实验数据表明,微管相关蛋白tau 和Aβ代谢可能干扰线粒体自噬机制[49-50]。有研究进一步发现,通过上调诱导转录因子EB (transcription factor EB,TFEB)的表达,将提高溶酶体蛋白的有效表达,关于线粒体自噬的介导标记物也会同步增加,由此可推测溶酶体功能缺陷可能会降低线粒体自噬,故溶酶体功能缺陷被认为是AD状态下线粒体自噬异常的重要机制之一[51]。上述证据可以推测线粒体自噬异常与微管相关蛋白tau、Aβ代谢及溶酶体功能缺陷存在密切关系;其通过干预微管相关蛋白tau 和Aβ代谢或溶酶体蛋白表达水平,提高线粒体自噬,能够进一步延缓AD的发生发展。
3 结语
AD发病机理复杂。现有研究表明,线粒体是AD发病进程中的关键环节之一,多个线粒体相关要素参与调控了AD的发展,其中包括线粒体能量代谢、线粒体Ca2+稳态、线粒体氧化应激、线粒体动力学、线粒体DNA、线粒体自噬等,针对线粒体功能障碍的靶向性治疗的基础研究取得了一定的进展,今后需继续从不同的层面深入研究线粒体功能障碍与AD之间的具体机制,明确线粒体异常与AD病理生理之间的关系,将有利于寻找线粒体的靶向防治策略,为临床上AD 的诊疗提供新思路及科学依据,对AD 的防治及延缓该疾病的进展有着重大的价值。
