抗菌新药zoliflodacin研究进展
2023-05-30陈虹彤李国庆游雪甫杨信怡
陈虹彤 李国庆 游雪甫 杨信怡
摘要:Zoliflodacin是螺嘧啶三酮类抗菌药,结构新颖,是一种新型细菌Ⅱ型拓扑异构酶抑制剂,其作用位点不同于氟喹诺酮类药物,目前正开发用于单纯性淋病的治疗。Zoliflodacin对包括多重耐药菌株在内的淋病奈瑟球菌具有良好的体外抗菌活性,单用zoliflodacin或与其他抗菌药物联用对淋病奈瑟球菌具有快速殺菌作用,对多种革兰阳性菌、革兰阴性苛养菌、非典型病原体也具有良好抗菌活性。实验室条件下,zoliflodacin诱导淋病奈瑟球菌及金黄色葡萄球菌发生自发耐药突变的频率较低(10-8~10-9)。已有研究中,未观察到zoliflodacin与氟喹诺酮类、大环内酯类、β-内酰胺类、糖肽类、恶唑烷酮类和四环素类抗菌药存在交叉耐药性。Ⅰ期临床试验显示zoliflodacin具有良好的口服生物利用度及较高的安全性;Ⅱ期临床试验表明zoliflodacin可有效治疗泌尿生殖道和直肠淋病奈瑟球菌感染,主要药物相关不良反应为胃肠道反应。目前,正在开展一项zoliflodacin的全球Ⅲ期临床试验研究。该药有望成为一种治疗耐药淋病奈瑟球菌感染的新型口服抗菌药,应用前景良好。
关键词:Zoliflodacin;药效学;药动学;临床应用;不良反应
中图分类号:R978.1文献标志码:A
Research progress of the new antibacterial drug zoliflodacin
Chen Hong-tong, Li Guo-qing, You Xue-fu, and Yang Xin-yi
(Beijing Key Laboratory of Antimicrobial Agents, Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050)
Abstract Zoliflodacin is a novel spiropyrimidinetrione antibacterial agent, which inhibits bacterial type Ⅱ topoisomerase at a unique site of action different from that of fluoroquinolones, and is currently being developed for the treatment of uncomplicated gonorrhea. Zoliflodacin has an excellent in vitro antibacterial activity against Neisseria gonorrhoeae including multi-drug resistant strains. It alone or in combination with other antibacterial drugs shows a rapid bactericidal effect on Neisseria gonorrhoeae. Zoliflodacin also shows good in vitro antibacterial activities against other Gram-positive bacteria, Gram-negative fastidious bacteria, and atypical pathogens. Under laboratory conditions, the frequency of spontaneous mutation of Neisseria gonorrhoeae and Staphylococcus aureus induced by zoliflodacin was low (10-8~10-9). There is no cross-resistance that has been observed between zoliflodacin and antibiotics including fluoroquinolones, macrolides, β-lactams, glycopeptides, oxazolidinones, and tetracyclines in previous studies. Zoliflodacin showed good oral bioavailability and high safety in healthy volunteers in phase I clinical trials. A phase Ⅱ clinical trial demonstrated that zoliflodacin could effectively treat gonococcal urogenital and rectal infections, and the main drug-related adverse reactions for this antibiotic were gastrointestinal problems. At present, a global phase Ⅲ clinical trial of zoliflodacin for the treatment of uncomplicated gonorrhoea is underway and the antibiotic is expected to become a new type of oral treatment option for drug-resistant Neisseria gonorrhoeae infections with good application prospects.
Key words Zoliflodacin; Pharmacodynamics; Pharmacokinetics; Clinical application; Adverse reactions
淋病由淋病奈瑟球菌(Neisseria gonorrhoeae)引起,是一种在世界范围内最常见的性传播疾病,据估计全球每年有8000万人感染[1]。在我国,淋病与病毒性肝炎、艾滋病、梅毒等同属乙类传染病。过去几十年间,磺胺类药物、青霉素、四环素、壮观霉素、环丙沙星或氧氟沙星、阿奇霉素、头孢菌素等都曾先后作为治疗淋病的“特效药”。然而,随着这些药物的持续使用,淋病奈瑟球菌已逐渐丧失对它们的敏感性。迄今,耐药甚至耐多药菌株在临床上已十分普遍,且呈逐年增多趋势,因此,寻找和开发新的有效治疗药物势在必行。
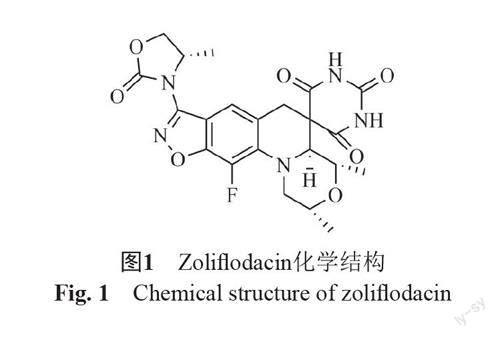
Zoliflodacin(AZD0914,ETX0914,唑利氟达星)是Entasis Therapeutics公司新开发的螺嘧啶三酮类抗菌药物,结构新颖,抗菌谱较广,是一类新型拓扑异构酶抑制剂的代表,现主要开发用于治疗无并发症的单纯性淋病。Zoliflodacin分子式为C22H22FN5O7(化学结构式见图1),分子量(MW)为487.44,主要通过抑制细菌Ⅱ型拓扑异构酶,导致细菌中DNA双链断裂的积累而发挥抗菌作用。该药结合位点异于氟喹诺酮类抗菌药,作用机制亦不同于迄今所用过的其他淋病治疗药物。本文重点围绕zoliflodacin最新临床前和临床研究进展,就该药的构效关系、作用机制、体内外抑杀菌活性、临床疗效等方面逐一综述,供抗感染研究领域医药工作者参考。
1 Zoliflodacin的发现与构效关系
Zoliflodacin是在一项旨在改善先導化合物PNU-286607(QPT-1,图2)的药物代谢与药动学(DMPK)和抗菌特性的定向优化工作中所获得。PNU-286607是第一个获得报道的螺嘧啶三酮类化合物,最早由法玛西亚普强公司(Pharmacia & Upjohn)在高通量筛选中发现。反向基因组学研究结果表明该化合物属于DNA促旋酶抑制剂,对革兰阳性菌和营养需求较高的革兰阴性苛养菌(如淋病奈瑟球菌)具有抗菌活性[2-3]。
此后,辉瑞公司的研究人员进一步探索了PNU-286607的构效关系,包括在苯环各位置引入不同取代基及对螺嘧啶三酮药效基团和吗啉环进行修饰。随后,阿斯利康研究人员又采用苯并异恶唑取代苯环,合成了一系列化合物,并对其中的化合物2(图 2)进行了活性测定。然而,化合物2因明显的骨髓毒性和遗传毒性未能进入临床试验[4]。但在进一步的构效关系研究中,Entasis Therapeutics公司的研究者发现,苯并异恶唑环上含有恶唑烷酮取代基的化合物zoliflodacin具有较高的抗菌活性,且骨架中引入的恶唑烷酮基团减轻了拓扑异构酶抑制剂中常见的毒性问题,避免了其他基团取代中常见的骨髓毒性和遗传毒性[5-6]。目前,zoliflodacin作为首个用于淋病治疗的螺嘧啶三酮类药物,进入到了Ⅲ期临床试验阶段。
2 作用机制及潜在耐药机制
在抗感染治疗领域,氟喹诺酮类抗菌药一直是应用最为广泛的拓扑异构酶抑制剂。然而,长期使用导致的细菌耐药问题,正日益限制该类药物的有效应用。螺嘧啶三酮作为一类新型细菌Ⅱ型拓扑异构酶抑制剂,具有全新的作用机制。研究显示,zoliflodacin与细菌的Ⅱ型拓扑异构酶的结合位点与氟喹诺酮类药物完全不同,可避免与目前临床常用抗菌药物的交叉耐药性[6]。
前期研究显示,zoliflodacin对DNA生物合成的IC50(half maximal inhibitory concentration,半抑制浓度)=0.037 μg/mL,对RNA生物合成的IC50=4.3 μg/mL,对蛋白质、细胞壁和脂肪酸生物合成的IC50>256 μg/mL,提示其主要作用机制是抑制细菌DNA生物合成。现研究则进一步揭示zoliflodacin可通过抑制细菌促旋酶催化的DNA超螺旋过程和拓扑异构酶Ⅳ催化的解连结(decatenation)过程,稳定酶-DNA复合物,这一作用特点与环丙沙星类似。DNA促旋酶是一种具有两个亚基的四异聚体(GyrA2-GyrB2),主要功能是调控细菌DNA的负超螺旋张力。已有报道表明,环丙沙星可通过相关的水分子,经非催化性镁离子与GyrA的天冬氨酸/谷氨酸(E88)和丝氨酸(S84)残基相互作用,稳定促旋酶-DNA复合体,从而抑制DNA促旋酶催化的DNA重新连接。Zoliflodacin不同于环丙沙星的是,从DNA-促旋酶-zoliflodacin复合物中去除镁离子后,并不会重新链接已发生断裂的DNA。据此,研究者认为zoliflodacin不是通过“水-金属离子桥梁”作用于GyrA亚基,而是作用于并不涉及镁离子螯合作用的GyrB残基[7]。对于GyrA发生氨基酸取代的环丙沙星耐药突变菌株,zoliflodacin依然具有良好的抗菌活性,MIC值较低,但对于GyrB发生氨基酸取代的突变菌株却具有较高的MIC值,而环丙沙星却对后者保持低MIC水平,这些关键数据均反映出zoliflodacin的作用机制有别于环丙沙星[7-8]。此外,还有研究显示zoliflodacin可诱导大肠埃希菌产生与环丙沙星水平相当的DNA损伤情况下的SOS效应,从而抑制对数生长期大肠埃希菌的DNA生物合成[7]。
淋病奈瑟球菌是一种适应性很强的病原体,可通过基因组的不断改变适应不利环境,并对抗菌药物产生多种耐药机制,已有的研究表明它们主要通过:①产酶破坏或修饰抗菌药物;②靶点修饰或保护以降低抗菌药的活性;③减少药物入胞;④增加药物外排[9]等方式获得耐药性。
从目前实验室条件下用zoliflodacin诱导的淋病奈瑟球菌耐药突变株中,检测到的均是位于GyrB上D429N、K450T或K450N位点的单氨基酸突变。这3个位置的突变,可致使药物对突变株的MIC水平升高至0.5~4 μg/mL[10-11]。由于zoliflodacin尚未正式上市,其在人体内的耐药突变方式尚不得而知[10]。但有报道提到,zoliflodacin对淋病奈瑟球菌的活性受细菌外排泵的明显影响。有研究者利用MtrCDE、MacAB和NorM外排泵表达上调的淋病奈瑟球菌突变株受试,发现在外排泵高表达的情况下,zoliflodacin对突变菌株OM-5和PM-4的MIC值从0.125 μg/mL分别上升至1和4 μg/mL[12]。GyrB的特异性突变可导致细菌对zoliflodacin的耐药性,而MtrCDE外排泵的失活则可完全恢复菌株对zoliflodacin的敏感性。在淋病奈瑟球菌H041菌株MacAB或NorM外排泵失活的条件下,可使该菌株对zoliflodacin的MIC由0.125 μg/mL降低至0.008 μg/mL。但目前已完成的临床研究中,研究者尚未观察到淋病奈瑟球菌临床菌株因外排泵高表达导致的对zoliflodacin敏感性降低[12]。
在其他研究中,研究者还发现细菌接种量、温度、大气环境(空气、5% CO2或厌氧环境)、二价阳离子、人血清、人尿液、肺表面活性剂等均不影响zoliflodacin的体外抗菌活性;pH在5~8范围内变化时不影响该药对金黄色葡萄球菌、粪肠球菌、大肠埃希菌的抗菌活性,但pH超过8时,MIC升高≥8倍。这一现象可能与pH升高时,该药物分子的脱质子化有关[13]。
3 临床前药理学
3.1 体外抑菌活性
Zoliflodacin对不同国家1995株淋病奈瑟球菌临床分离株的MIC测定结果见表1。对受试的淋病奈瑟球菌,zoliflodacin的MIC90均≤0.25 μg/mL,对绝大多数受试菌的MIC90值为0.125 μg/mL。Zoliflodacin对近年临床分离的氟喹诺酮类耐药株及包括大环内酯类、β-内酰胺类、四环素在内的耐药株均具良好活性,也反映出该药与这些药物不存在交叉耐药性[14-17]。其中,zoliflodacin对环丙沙星耐药淋病奈瑟球菌的MIC90=0.06~0.125 μg/mL,对环丙沙星及头孢曲松双重耐药株的MIC=0.06 μg/mL[17]。同时,其对耐多药淋病奈瑟球菌也具有显著的抑菌作用,MIC90=0.25 μg/mL。
Zoliflodacin对临床分离的革兰阳性菌、其他革兰阴性菌、厌氧菌的MIC测定结果见表2。总体而言,其对革兰阳性菌及革兰阴性苛养菌具有良好的体外抗菌活性。其中,对金黄色葡萄球菌(11680株)、凝固酶阴性葡萄球菌(1923株)、链球菌(4380株)、卡他莫拉菌(145株)的MIC90=0.25 μg/mL,对路邓葡萄球菌(120株)、流感嗜血桿菌(352株)的MIC90=0.5 μg/mL,对粪肠球菌(1241株)的MIC90=1 μg/mL,对副流感嗜血杆菌(70株)的MIC90=2 μg/mL,但对屎肠球菌(946株)的抗菌活性稍弱,MIC90=8 μg/mL。Zoliflodacin的抗菌谱中涵盖了耐甲氧西林葡萄球菌、耐青霉素链球菌、耐万古霉素肠球菌、产β-内酰胺酶的嗜血杆菌属、耐氟喹诺酮卡他莫拉菌[20]。
从表2中可见,zoliflodacin对皮肤软组织感染、呼吸系统感染、全身性感染中常见的金黄色葡萄球菌(包括甲氧西林耐药、万古霉素中介、万古霉素耐药株)、凝固酶阴性葡萄球菌、链球菌属(包括无乳链球菌、肺炎链球菌、化脓性链球菌)等革兰阳性菌的抗菌活性较突出(MIC90=0.25~0.5 μg/mL),且对受试的红霉素、左氧氟沙星、利奈唑胺和/或万古霉素耐药菌株均具抗菌活性,在喹诺酮类耐药菌株中均未观察到交叉耐药性[21]。
Zoliflodacin对鲍曼不动杆菌、大肠埃希菌、肺炎克雷伯菌、铜绿假单胞菌等临床常见革兰阴性菌的抗菌活性较弱,MIC90分别为32、4、128μg/mL和>64 μg/mL。其对大肠埃希菌ATCC25922 (tolC)和铜绿假单胞菌PAO1(mexABCDXY)外排泵突变株的MIC值是相应野生型菌株的32倍,提示外排泵可能在革兰阴性菌对zoliflodacin的耐药中发挥着重要作用[21]。对于受试的厌氧菌,zoliflodacin对艰难梭菌MIC=0.125 μg/mL;但对拟杆菌活性相对较低,MIC=2~8 μg/mL[21]。
Zoliflodacin对包括泌尿生殖系统感染相关病原体在内的多种非典型病原菌亦具有良好的体外抗菌活性,结果见表3。Zoliflodacin对沙眼衣原、肺炎衣原体的MIC90分别为0.25 μg/mL(0.06~0.5 μg/mL)和1 μg/mL
(0.25~1 μg/mL),MBC90分别为0.5 μg/mL(0.125~
1 μg/mL)和2 μg/mL(0.5~2 μg/mL)[22]。其对生殖支原体和肺炎支原体的活性(MIC90均为1 μg/mL)与左氧氟沙星和多西环素相当;对脲原体属的IC90(1 μg/mL)是阿奇霉素的1/4,左氧氟沙星和多西环素的1/8;但对人支原体的MIC90(4 μg/mL)是左氧氟沙星的8倍[23]。
3.2 体外杀菌活性
有关zoliflodacin体外杀菌活性的研究显示,其对包括氟喹诺酮耐药株在内的7株受试金黄色葡萄球菌和1株受试化脓性链球菌均表现出体外杀菌活性,MBC(最小杀菌浓度)在2倍MIC浓度范围内。时间-杀菌曲线测定结果显示,≥2×MIC浓度的zoliflodacin作用6 h内,可使金黄色葡萄球菌USA100(左氧氟沙星耐药)和肺炎链球菌ATCC 49619降低≥3 log级[21]。8×MIC和16×MIC的zoliflodacin对淋病奈瑟球菌显示出浓度依赖性的快速杀菌作用,在2×MIC和4×MIC浓度下杀菌作用则较慢[11]。
3.3 抗生素后效应(postantibiotic effects,PAE)
在1×MIC、4×MIC、16×MIC浓度下,zoliflodacin对金黄色葡萄球菌USA100和肺炎链球菌ATCC 49619的PAE范围分别为0.25~0.55 h、1.65~2.4 h、2.15~>3.3 h,和以往报道的喹诺酮类及氟喹诺酮类药物对金黄色葡萄球菌的PAE相近[21, 24-25]。
3.4 联合抑菌活性
有研究者采用棋盘法,根据分数抑制浓度指数(fractional inhibitory concentration index,FICI)对zoliflodacin与多种抗菌药物的联用效应进行了评价。结果显示,zoliflodacin与绝大多数抗菌药物联用时,对多数淋病奈瑟球菌菌株表现为无关效应(FICI=0.5~3)。但zoliflodacin与环丙沙星联用对野生株WHO F、zoliflodacin耐药突变株OM-5、PM-4具有协同抗菌效应(FICI=0.5、FICI=0.4~1);与头孢曲松联用对野生株WHO P及突变株PM-4具有协同抗菌效应(FICI=0.5~0.6)[12]。时间-杀菌曲线显示,与单用zoliflodacin相比,zoliflodacin与环丙沙星或阿奇霉素联用产生更快的杀菌效果。采用耐药突变株进行测试时,联用上述药物后,突变株的最小生长速率(ψmin)也显著低于单用zoliflodacin时的情况[12]。另一时间-杀菌曲线的研究报道表明,zoliflodacin与四环素、喹红霉素、头孢曲松或庆大霉素联用均可增强zoliflodacin对淋病奈瑟球菌的杀伤作用[11]。Zoliflodacin与受试抗菌药物联用时均未产生拮抗作用。
3.5 自发突变率
相较于氟喹诺酮类药物,金黄色葡萄球菌(5株)和淋病奈瑟球菌(5株)对zoliflodacin诱导所产生的耐药突变频率较低(4×MIC作用下自发突变率分别为<1.1×10-9,1.5×10-8~<5.2×10-9)。同时,zoliflodacin自发耐药突变体对氟喹诺酮类药物不显示交叉耐药性[6,8]。这些突变体对环丙沙星、新生霉素和NBTI(氨基哌啶类代表化合物)完全敏感,进一步证实了zoliflodacin不同于其他细菌拓扑异构酶抑制剂的作用模式。
4 药理学及PK/PD研究
Zoliflodacin在临床前实验动物研究中PK参数见表4。相较于小鼠和大鼠,犬和猴子对zoliflodacin有较高的生物利用度,表明zoliflodacin首过效应较低。犬和猴子口服或静脉注射zoliflodacin的血浆半衰期相近,表明在这些物种中zoliflodacin的吸收不是限速过程。小鼠、大鼠、犬和人体[14C]-zoliflodacin血浆蛋白结合率相近(17%~22.1%),且与zoliflodacin浓度(1~50 ?mol/L)无相关性。
小鼠、大鼠、犬和人肝细胞对zoliflodacin的体外生物转化研究显示,相较于小鼠和大鼠,该药在犬和人肝细胞中的内在清除率较低。从大鼠静脉注射 [14C]-zoliflodacin后,尿液、胆汁和粪便中药物的平均回收率分别约为给药剂量的15%、55%和24%。在尿液和胆汁中回收的原型药低于给药剂量的5%,这表明zoliflodacin在大鼠体内经历了广泛的代谢过程。在大鼠和小鼠尿液中检测到的细胞色素P450(CYP酶)介导的代谢产物M1、M2、M4、M6和M12,在预先给予1-氨基苯并三唑(一种细胞色素P450酶非特异性抑制剂)的小鼠尿液样本中均未检出,这提示zoliflodacin是CYP酶的有效代谢底物。在无菌条件下将zoliflodacin与无菌大鼠新鲜粪便共孵育,或与大鼠肠道微粒体和细胞质孵育时,不形成在普通大鼠粪便中检测到的主要代谢物,这表明zoliflodacin很可能直接排泄到肠腔中,由肠道菌群介导部分代谢产物的生成。Zoliflodacin肝脏代谢的主要位点包括CYP酶介导的吗啉环的氧化和恶唑烷酮环的修饰,而苯异恶唑环的还原主要发生在肠道中[26]。
用于确定PK/PD和评估临床剂量方案的体外动态模型系统如中空纤维感染模型(hollow fiber infection model,HFIM),直到近年才被成功应用于淋病奈瑟球菌的治疗评价[27]。基于淋病奈瑟球菌HFIM体外动态感染模型,研究人员对zoliflodacin治疗淋病奈瑟球菌参考菌株WHO F(对所有相关抗菌药物敏感)和WHO X(对包括头孢曲松在内的抗菌药物广泛耐药)展开了药效学(7 d内)评价。研究者评价了不同给药方案[全剂量给药1次、半剂量给药2次(q12 h)、1/3剂量给药3次(q8 h)]对淋病奈瑟球菌WHO X的杀菌效应。结果如图3所示,zoliflodacin对受试菌株主要表现为浓度依赖性杀菌作用,而非时间依赖性作用关系。全剂量单次给药条件下,细菌菌落计数下降速度最快;采用较大给药剂量(>2 g)时,杀菌效果与给药剂量、次数的相关性较小。>2 g zoliflodacin对淋病奈瑟球菌的杀菌效应可达到最大杀灭速率。对于 WHO F 和 WHO X感染,单次口服zoliflodacin 2 g对应的AUC/MIC分别为137.6 和75.6[28]。而要有效杀灭淋病奈瑟球菌并抑制耐药性的出现,则应采用>2 g的单次口服剂量给药。此外,目前只有人体血浆游离zoliflodacin PK數据可用于PK/PD评价,基于这一PK数据的淋病奈瑟球菌HFIM模型可能无法理想地反映该药在感染部位的PK/PD。泌尿生殖系统和生殖器外感染部位尤其是咽部的PK数据十分重要,有利于进一步优化zoliflodacin剂量预测的准确性[28]。
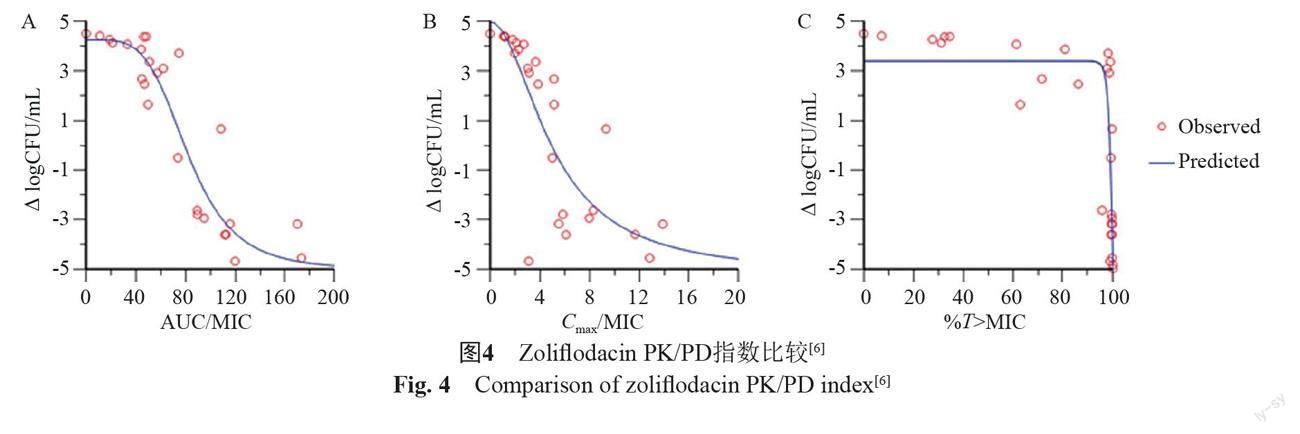
由于zoliflodacin的作用机制与其他已成功应用于淋病治疗的拓扑异构酶抑制剂相似,替代病原体方法被用于zoliflodacin的PK/PD评估。人们利用甲氧西林敏感金黄色葡萄球菌ARC516的HFIM定义zoliflodacin PK/PD指数发现,与其他细菌拓扑异构酶抑制剂类似,AUC/MIC是和zoliflodacin活性最相关的PK/PD指标,相关系数(R2)=0.95,而Cmax/MIC、%T>MIC的R2分别为0.88和0.85(图4)[6]。
由于淋病奈瑟球菌在人体的独特适应性、淋病动物模型对淋病奈瑟球菌的自发清除特性,已有动物模型仅能提供该病原的定性反应结果而无法实现临床结果定量转化。目前的实验动物研究中,淋病奈瑟球菌的PK/PD治疗靶点尚待开发或建立,用已有动物模型评估和反映zoliflodacin治疗时的实用价值很不可靠[29]。为评估zoliflodacin对人体淋病奈瑟球菌的有效水平,研究者采用病原体替代方法,利用金黄色葡萄球菌导致中性粒细胞减少症的CD-1小鼠腿部感染模型对zoliflodacin的PK/PD进行测定。目前,该模型已被证明与人类临床淋病治疗的疗效相关[6,30]。利用PK/PD目标评估zoliflodacin在人体的有效水平,该小鼠大腿模型中金黄色葡萄球菌的平均fAUC/MIC为66(范围43~98)。结合zoliflodacin对淋病奈瑟球菌MIC90(0.12 μg/mL)和人体fu(17%),转化为预测的人体内有效平均AUC为49 μg·h/mL[6]。
5 安全性评价
为评价zoliflodacin对细菌拓扑异构酶的选择性活性,研究者测定了其对纯化的人Ⅱ型拓扑异构酶DNA的抑制活性。结果显示,zoliflodacin对人体Ⅱ型拓扑异构酶α(IC50>400 μmol/L)和β(IC50=79 μmol/L)的抑制作用很小,证明了zoliflodacin的高选择性[7]。
临床中,应用某些氟喹诺酮类药物和利奈唑胺时,会出现血小板减少症,而在zoliflodacin的实验研究中,未见其对血小板计数产生显著影响[31-32]。此外,一些氟喹诺酮类药物由于与hERG K+通道的结合,会导致QT间期延长,引起心律失常和尖端扭转型室性心动过速,如氟喹诺酮类药物司帕沙星和格雷沙星就曾因导致QT延长而被撤市。相比之下,zoliflodacin对包括hERG在内的心肌细胞动作电位相关的各种离子通道均显示出较高的IC50值,这表明zoliflodacin诱发心律失常的可能性较低[6]。
此外,研究者对zoliflodacin展开了多项临床前体内毒理学研究[6]。给予Wistar大鼠和Beagle犬zoliflodacin 28 d,收集的数据涵盖临床观察、体重变化、食物消耗量、临床病理学和组织病理学结果。在大鼠中,zoliflodacin口服耐受剂量为1000 mg/(kg·day)
(28 d)。其在大鼠中的无可见不良作用水平(no observable adverse effect level,NOAEL)为200 mg/kg,与第27天测得的 Cmax(107 μg/mL)和 AUC24 h(1070 μg·h/mL)
相关。大鼠fAUC是小鼠大腿模型中观察到的有效fAUC的13倍。在犬模型中,受试犬可耐受该药28 d
500 mg/(kg·day)的剂量(口服强饲给药)。在这一最高测定剂量下,可观察到动物心动过速,这与静脉遥测犬研究的结果一致,其在100 mg/kg静脉给药时可观察到血压、收缩力和PR间期降低及心率加快,但这些影响均短暂且完全可逆,相关反应可以在临床环境中进行有效监测。其他心电图参数(包括QTc或QRS)在zoliflodacin给药后未见变化。受试犬接受药物后,NOAEL为100 mg/kg(口服),第27天监测的Cmax为
85 μg/mL,AUC24h为870 μmol·h/mL。大鼠和犬在接受NOAEL水平的给药时,指示贫血或血小板减少症的血液学指标或血小板计数均无显著变化。总之,临床前体内毒性数据有力地支持了Entasis Therapeutics公司向美国FDA提交zoliflodacin的临床研究申请,启动健康志愿者单剂量研究,进而支持该药单剂量口服治疗单纯性淋病的临床试验[6]。
6 临床试验研究
6.1 Ⅰ期临床试验
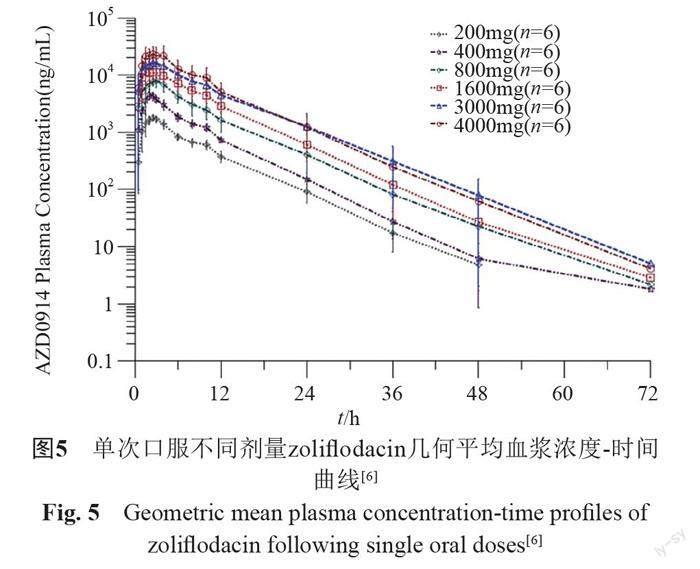
Ⅰ期临床试验NCT01929629评价了单次递增口服剂量(single ascending oral doses,SAD)以及进食和禁食状态下zoliflodacin的安全性、耐受性、药物代谢动力学(表5)。其中,在随机、双盲、安慰剂对照的单次递增剂量SAD试验中,受试者在禁食状态下(给药前禁食至少10 h至给药后4 h)接受单次口服剂量200、400、800、1600、3000和4000 mg zoliflodacin混悬液或安慰剂。各剂量zoliflodacin均吸收迅速,血浆药物浓度呈单相下降(图5),血清中药物达到最大浓度的时间(Tmax)为1.5~2.3 h。各浓度药物消除半衰期(t1/2)相对一致,范围在5.3~6.3 h。8名受试者和10名受试者分别接受两次zoliflodacin 1500 mg及3000 mg的单一口服剂量,且分别于进食或禁食状态进行评估。受试者在禁食状态下,Tmax=2.5 h;在进食状态下药物吸收延迟,Tmax=4 h。此外,在进食状态下,药物在1500及3000 mg剂量下测得的浓度-时间曲线下面积(AUC)均有所增加[33]。
另一Ⅰ期临床试验NCT02298920评估了健康志愿者单次口服3000 mg zoliflodacin后的吸收、分布、代谢和排泄(ADME)规律,6名男性受试者被纳入并完成了相关研究。AUC和Cmax参数与前一项递增剂量研究中的PK参数相近。97.8%的zoliflodacin随尿液(18.2%)及粪便(79.6%)排出。Zoliflodacin主要的清除途径是通过代谢并经粪便清除,代谢形式药物约为粪便排泄剂量的56%[33]。氧化代谢和还原代谢在zoliflodacin的體内消除中均发挥着重要作用。在上述Ⅰ期临床试验中,受试者均未见严重不良反应,轻度和非严重不良反应包括短暂味觉障碍和头痛等,所有受试者中出现的不良反应均可逆。
为评价zoliflodacin的心脏安全性,在72名健康受试者中开展了全面的QT/QTc(TQT)研究(NCT03613649)[34]。在该随机、双盲、安慰剂对照、四周期交叉(4-period crossover study)Ⅰ期临床研究中,受试者在禁食状态下分别接受单剂量口服2 g zoliflodacin、4 g zoliflodacin、安慰剂或400 mg莫西沙星,通过Fridericia公式校正的QT 间期(QTcF)持续时间来测量心脏复极化。Zoliflodacin对心率、PR间期、QRS间期、心电图形态和临床实验室检测(血液学、血清化学、尿液分析)没有临床显著影响。该试验表明zoliflodacin不致心律失常,证实了单剂量口服zoliflodacin的心脏安全性。
6.2 Ⅱ期临床试验
Ⅱ期临床试验NCT02257918评价了单剂量口服2 g或3 g zoliflodacin及肌肉注射500 mg头孢曲松治疗无并发症泌尿生殖道淋病患者的安全性及有效性,主要疗效指标是微生物学意向治疗(micro-ITT)人群中泌尿生殖系统的微生物学治愈比例,结果见表6。共179名患者(167名男性及12名女性)被纳入该项研究,在共144名可评估的micro-ITT参与者中,接受2 g zoliflodacin、3 g zoliflodacin的受试者泌尿生殖部位的微生物学治愈率分别为96%(55/57)和96%(54/56)。所有5名接受2 g zoliflodacin、7名接受3 g zoliflodacin以及3名接受头孢曲松组的直肠感染患者均获成功治愈。接受2 g zoliflodacin、3 g zoliflodacin和500 mg头孢曲松治疗的咽部感染受试者中,治愈率分别为50%(4/8)、82%(9/11)和100%(4/4)。研究中共报道了84例不良反应事件,21例为药物相关不良反应,其中绝大多数为胃肠道反应。大多数无并发症的泌尿生殖道和直肠淋病球菌感染患者在采用口服zoliflodacin后获成功治愈,但zoliflodacin在治疗咽部感染方面疗效欠佳[35]。治疗未愈患者的咽部分离菌中,未检测出耐药性或治疗后药物敏感性的显著变化,推测大多数咽部治疗失败的原因可能是zoliflodacin穿透咽部组织能力较差所致,不是因为再感染或耐药性病原体所引起[36]。
6.3 Ⅲ期临床试验
目前,Entasis Therapeutics公司与“全球抗生素研发伙伴”组织(The Global Antibiotic Research and Development Partnership,GARDP)合作,一项多中心、开放标签、随机对照、非劣效Ⅲ期临床试验NCT03959527正在招募志愿者,该临床研究预计将包括来自美国、荷兰、泰国和南非的约1000名泌尿生殖道淋病成人患者。该试验将对联用单次肌肉注射500 mg头孢曲松和单次口服1 g阿奇霉素与单纯口服3 g zoliflodacin治疗单纯性淋病进行比较性评价[37]。与Ⅱ期临床试验相同,该试验将淋病奈瑟球菌引起的泌尿生殖道感染作为纳入的标准。此外,泌尿生殖道外感染将作为次要终点进行研究,以帮助研究者了解zoliflodacin对这些感染的疗效。
7 展望
淋病及淋病奈瑟球菌耐药菌株的流行一直是广受关注的全球公共卫生问题。在世界范围内,细菌耐药性的发展既给淋病的有效治疗带来巨大的挑战,亦对淋病严重并发症的控制和治疗成本造成巨大负担[9]。淋病奈瑟球菌的耐药性进化数据显示,在抗生素开始应用之前,除penB外,从淋病奈瑟球菌中未检出过其他耐药性元件,目前的耐药性是由于抗菌药物的广泛应用或滥用导致[38]。由于过去几十年中,淋病奈瑟球菌对青霉素、四环素、壮观霉素、头孢克肟和氟喹诺酮类等药物不断产生耐药性,已使得可用的治疗选择日益减少[9]。
目前临床治疗淋病的推荐方案是采用头孢曲松与阿奇霉素联用,但世界范围内又逐渐出现了头孢曲松/阿奇霉素的耐药分离株。据统计,在2013—2016年期间,从我国临床分离的淋病奈瑟球菌中,有近20%对阿奇霉素耐药、10%对头孢曲松敏感性较低[39]。而在2018年,英国和澳大利亚发现了首批既对头孢曲松耐药又对阿奇霉素高水平耐药的淋病奈瑟球菌分离株[40-41]。为应对这一威胁,各国疾病控制与预防中心和世界卫生组织将耐第三代头孢菌素和氟喹诺酮的淋病奈瑟球菌作为紧急威胁的第二优先级病原体[42]。在我国,淋病奈瑟球菌耐药已被国家卫健委合理用药专家委员会列为耐药监测的重点类别,与全国14个政府部门联合发布的《遏制细菌耐药国家行动计划》的启动相呼应[43-44]。基于上述现状,开发有效治疗生殖道和生殖器外淋病的新型抗菌药物在应对这一全球公共卫生威胁方面尤为重要,也十分迫切。
为应对耐药淋病奈瑟球菌的治疗需求,zoliflodacin作为新型抗菌药物的代表品种,基于新型苯并异恶唑结构骨架设计,含有螺嘧啶三酮药效基团,是首个该类结构的抗菌药物。目前的研究已证明zoliflodacin对包括环丙沙星耐药菌株和头孢曲松耐药菌株在内的耐药淋病奈瑟球菌,以及包括沙眼衣原體和生殖支原体在内的其他性传播病原体都具有疗效,可用于单纯性淋病的治疗。Zoliflodacin作为一种口服抗菌药,通过抑制细菌Ⅱ型拓扑异构酶发挥杀菌作用。其独特的作用机制,使其和其他抗菌药物没有交叉耐药性,因而可有效治疗氟喹诺酮耐药淋病奈瑟球菌引发的感染,Ⅱ期临床试验证实的zoliflodacin安全性和有效性也进一步支持了这一结论。总之,目前的实验结果表明该药有望成为治疗淋病的新型口服药物。
此外,有研究者指出,目前淋病治疗新指南的制定通常只考虑某种治疗对淋病奈瑟球菌的耐药性诱导作用。然而,淋病奈瑟球菌的耐药性往往首先出现在共生的其他奈瑟菌中,再通过相关耐药性基因的水平转移传递给淋病奈瑟球菌。这提示在zoliflodacin未来的临床试验中应加强监测,着力关注人体内共生奈瑟菌对该药敏感性的变化[45]。
参 考 文 献
Wilson M E. Antibiotics: What Everyone Needs to Know?[M]. New York: Oxford University Press, 2019: 432.
Miller A A, Bundy G L, Mott J E, et al. Discovery and characterization of QPT-1, the progenitor of a new class of bacterial topoisomerase inhibitors[J]. Antimicrob Agents Chemother, 2008, 52(8): 2806-2812.
Bradford P A, Miller A A, ODonnell J, et al. Zoliflodacin: an oral spiropyrimidinetrione antibiotic for the treatment of Neisseria gonorrhoeae, including multi-drug-resistant isolates[J]. ACS Infect Dis, 2020, 6(6): 1332-1345.
Basarab G S, Brassil P, Doig P, et al. Novel DNA gyrase inhibiting spiropyrimidinetriones with a benzisoxazole scaffold: SAR and in vivo characterization[J]. J Med Chem, 2014, 57(21): 9078-9095.
Shi C, Zhang Y, Wang T, et al. Design, synthesis, and biological evaluation of novel DNA gyrase-inhibiting spiropyrimidinetriones as potent antibiotics for treatment of infections caused by multidrug-resistant gram-positive bacteria[J]. J Med Chem, 2019, 62(6): 2950-2973.
Basarab G S, Kern G H, McNulty J, et al. Responding to the challenge of untreatable gonorrhea: ETX0914, a first-in-class agent with a distinct mechanism-of-action against bacterial Type Ⅱ topoisomerases[J]. Sci Rep, 2015, 5(1): 11827.
Kern G, Palmer T, Ehmann D E, et al. Inhibition of Neisseria gonorrhoeae Type Ⅱ Topoisomerases by the Novel Spiropyrimidinetrione AZD0914[J]. J Biol Chem, 2015, 290(34): 20984-20994.
Alm R A, Lahiri S D, Kutschke A, et al. Characterization of the novel DNA gyrase inhibitor AZD0914: Low resistance potential and lack of cross-resistance in Neisseria gonorrhoeae[J]. Antimicrob Agents Chemother, 2015, 59(3): 1478-1486.
Unemo M, Shafer W M. Antimicrobial resistance in Neisseria gonorrhoeae in the 21st century: Past, evolution, and future[J]. Clin Microbiol Rev, 2014, 27(3): 587-613.
Adamson P C, Lin E Y, Ha S M, et al. Using a public database of Neisseria gonorrhoeae genomes to detect mutations associated with zoliflodacin resistance[J]. J Antimicrob Chemother, 2021, 76(11): 2847-2849.
Foerster S, Drusano G, Golparian D, et al. In vitro antimicrobial combination testing of and evolution of resistance to the first-in-class spiropyrimidinetrione zoliflodacin combined with six therapeutically relevant antimicrobials for Neisseria gonorrhoeae[J]. J Antimicrob Chemother, 2019, 74(12): 3521-3529.
Foerster S, Golparian D, Jacobsson S, et al. Genetic resistance determinants, in vitro time-kill curve analysis and pharmacodynamic functions for the novel topoisomerase Ⅱ inhibitor ETX0914 (AZD0914) in Neisseria gonorrhoeae[J]. Front Microbiol, 2015, 6: 1377.
Giacobbe R A, Huband M D, DeJonge B L, et al. Effect of susceptibility testing conditions on the in vitro antibacterial activity of ETX0914[J]. Diagn Microbiol Infect Dis, 2017, 87(2): 139-142.
Su X H, Wang B X, Le W J, et al. Multidrug-resistant Neisseria gonorrhoeae isolates from nanjing, China, are sensitive to killing by a novel DNA gyrase inhibitor, ETX0914 (AZD0914)[J]. Antimicrob Agents Chemother, 2016, 60(1): 621-623.
Papp J R, Lawrence K, Sharpe S, et al. In vitro growth of multidrug-resistant Neisseria gonorrhoeae isolates is inhibited by ETX0914, a novel spiropyrimidinetrione[J]. Int J Antimicrob Agents, 2016, 48(3): 328-330.
Unemo M, Ringlander J, Wiggins C, et al. High In vitro susceptibility to the novel spiropyrimidinetrione ETX0914 (AZD0914) among 873 contemporary clinical Neisseria gonorrhoeae isolates from 21 European countries from 2012 to 2014[J]. Antimicrob Agents Chemother, 2015, 59(9): 5220-5225.
Azam M A, Thathan J, and Jubie S. Dual targeting DNA gyrase B (GyrB) and topoisomerse Ⅳ (ParE) inhibitors: A review[J]. Bioorg Chem, 2015, 62: 41-63.
Jacobsson S, Golparian D, Alm R A, et al. High in vitro activity of the novel spiropyrimidinetrione AZD0914, a DNA gyrase inhibitor, against multidrug-resistant Neisseria gonorrhoeae isolates suggests a new effective option for oral treatment of gonorrhea[J]. Antimicrob Agents Chemother, 2014, 58(9): 5585-5588.
Jacobsson S, Kularatne R, Kittiyaowamarn R, et al. High in vitro susceptibility to the first-in-class spiropyrimidinetrione zoliflodacin among consecutive clinical Neisseria gonorrhoeae isolates from Thailand (2018) and South Africa (2015-2017)[J/OL]. Antimicrob Agents Chemother, 2019, 63(12). [2021-10-05]. https://journals.asm.org/doi/full/10.1128/AAC.01479-19.
Biedenbach D J, Huband M D, Hackel M, et al. In vitro activity of AZD0914, a novel bacterial DNA gyrase/topoisomerase Ⅳ inhibitor, against clinically relevant Gram-positive and fastidious gram-negative pathogens[J]. Antimicrob Agents Chemother, 2015, 59(10): 6053-6063.
Huband M D, Bradford P A, Otterson L G, et al. In vitro antibacterial activity of AZD0914, a new spiropyrimidinetrione DNA gyrase/topoisomerase inhibitor with potent activity against Gram-positive, fastidious Gram-Negative, and atypical bacteria[J]. Antimicrob Agents Chemother, 2015, 59(1): 467-474.
Kohlhoff S A, Huband M D, and Hammerschlag M R. In vitro activity of AZD0914, a novel DNA gyrase inhibitor, against Chlamydia trachomatis and Chlamydia pneumoniae[J]. Antimicrob Agents Chemother, 2014, 58(12): 7595-7596.
Waites K B, Crabb D M, Duffy L B, et al. In vitro antibacterial activity of AZD0914 against human Mycoplasmas and Ureaplasmas[J]. Antimicrob Agents Chemother, 2015, 59(6): 3627-3629.
Spangler S K, Bajaksouzian S, Jacobs M R, et al. Postantibiotic effects of grepafloxacin compared to those of five other agents against 12 Gram-positive and -negative bacteria[J]. Antimicrob Agents Chemother, 2000, 44(1): 186-189.
Pankuch G A and Appelbaum P C. Postantibiotic effect of DX-619 against 16 gram-positive organisms[J]. Antimicrob Agents Chemother, 2005, 49(9): 3963-3965.
Guo J, Joubran C, Luzietti R A, et al. Absorption, distribution, metabolism and elimination of 14C-ETX0914, a novel inhibitor of bacterial type-Ⅱ topoisomerases in rodents[J]. Xenobiotica, 2017, 47(1): 31-49.
VanScoy B D, Fikes S, Bhavnani S M, et al. A Hollow-fiber infection model to evaluate the prevention of on-therapy resistance of Neisseria gonorrhoeae to gepotidacin[R]. Washington, DC: STD Prevention Conference, 2018.
Jacobsson S, Golparian D, Oxelbark J, et al. Pharmacodynamic evaluation of dosing, bacterial kill, and resistance suppression for zoliflodacin against Neisseria gonorrhoeae in a dynamic hollow fiber infection model[J]. Front Pharmacol, 2021, 12: 682135.
Jerse A E, Wu H, Packiam M, et al. Estradiol-treated female mice as surrogate hosts for Neisseria gonorrhoeae genital tract infections[J]. Front Microbiol, 2011, 2: 107.
Craig W A. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men[J]. Clin Infect Dis, 1998, 26(1): 1-12.
Attassi K, Hershberger E, Alam R, et al. Thrombocytopenia associated with linezolid therapy[J]. Clin Infect Dis, 2002, 34(5): 695-698.
Cheah C Y, De Keulenaer B, and Leahy M F. Fluoroquinolone-induced immune thrombocytopenia: A report and review[J]. Intern Med J, 2009, 39(9): 619-623.
ODonnell J, Lawrence K, Vishwanathan K, et al. Single-dose pharmacokinetics, excretion, and metabolism of zoliflodacin, a novel spiropyrimidinetrione antibiotic, in healthy volunteers[J/OL]. Antimicrob Agents Chemother, 2019, 63(1): e01808-18. [2021-10-05]. https://journals.asm.org/doi/full/10.1128/AAC.01808-18.
Newman L M, Kankam M, Nakamura A, et al. Thorough QT study to evaluate the effect of zoliflodacin, a novel therapeutic for gonorrhea, on cardiac repolarization in healthy adults[J]. Antimicrob Agents Chemother, 2021, 65(12): e0129221.
Taylor S N, Marrazzo J, Batteiger B E, et al. Single-dose zoliflodacin (ETX0914) for treatment of urogenital gonorrhea[J]. N Engl J Med, 2018, 379(19): 1835-1845.
Manavi K, Young H, and McMillan A. The outcome of oropharyngeal gonorrhoea treatment with different regimens[J]. Int J STD AIDS, 2005, 16(1): 68-70.
Global antibiotics research and development partnership. zoliflodacin in uncomplicated gonorrhoea[EB/OL]. https://clinicaltrials.gov/ct2/show/NCT03959527?term=Zoliflodacin&draw=2&rank=3, 2021-09-21/2021-10-05.
Golparian D, Harris S R, Sánchez-Busó L, et al. Genomic evolution of Neisseria gonorrhoeae since the preantibiotic era (1928-2013): Antimicrobial use/misuse selects for resistance and drives evolution[J]. BMC Genomics, 2020, 21(1): 116.
Yin Y P, Han Y, Dai X Q, et al. Susceptibility of Neisseria gonorrhoeae to azithromycin and ceftriaxone in China: A retrospective study of national surveillance data from 2013 to 2016[J]. PLoS Med, 2018, 15(2): e1002499.
Eyre D W, Sanderson N D, Lord E, et al. Gonorrhoea treatment failure caused by a Neisseria gonorrhoeae strain with combined ceftriaxone and high-level azithromycin resistance, England, February 2018[J]. Euro Surveill, 2018, 23(27): 2-7.
Whiley D M, Jennison A, Pearson J, et al. Genetic characterisation of Neisseria gonorrhoeae resistant to both ceftriaxone and azithromycin[J]. Lancet Infect Dis, 2018, 18(7): 717-718.
Bloom D and Cadarette D. Infectious disease threats in the twenty-first century: strengthening the global response[J]. Front Immunol, 2019, 10: 549.
Xiao Y, Li L. China's national plan to combat antimicrobial resistance[J]. Lancet Infect Dis, 2016, 16(11): 1216-1218.
Chen X S, Yin Y P, Li X Y. A ROADMAP plan to address research needs for gonococcal antimicrobial resistance in China[J]. Clin Infect Dis, 2019, 68(3): 505-510.
Kenyon C, Laumen J, Manoharan-Basil S. choosing new therapies for gonorrhoea: we need to consider the impact on the pan-neisseria genome. A viewpoint[J/OL]. Antibiotics (Basel), 2021, 10(5): 515. [2021-10-05]. https://www.mdpi.com/2079-6382/10/5/515/htm.
收稿日期:2021-11-12
作者簡介:陈虹彤,女,生于1995年,在读博士研究生,主要从事抗微生物药物药理学及药物作用机制、耐药机制研究,E-mail: cht1010@163.com
通讯作者,E-mail:yangxinyi1976@hotmail.com
